
Selenium in Peptide Chemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Selenium and Sulfur Comparison of Properties and Reactivity
3. Selenium-Mediated Native Chemical Ligation
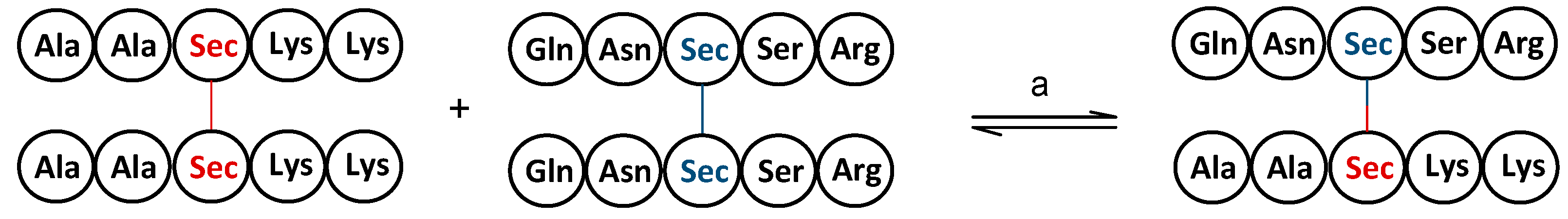
4. Redox Properties of Selenopeptides—Selective Formation of Se-Se Bridges in the Presence of S-H Groups
5. Identification and Detection of Selenopeptides
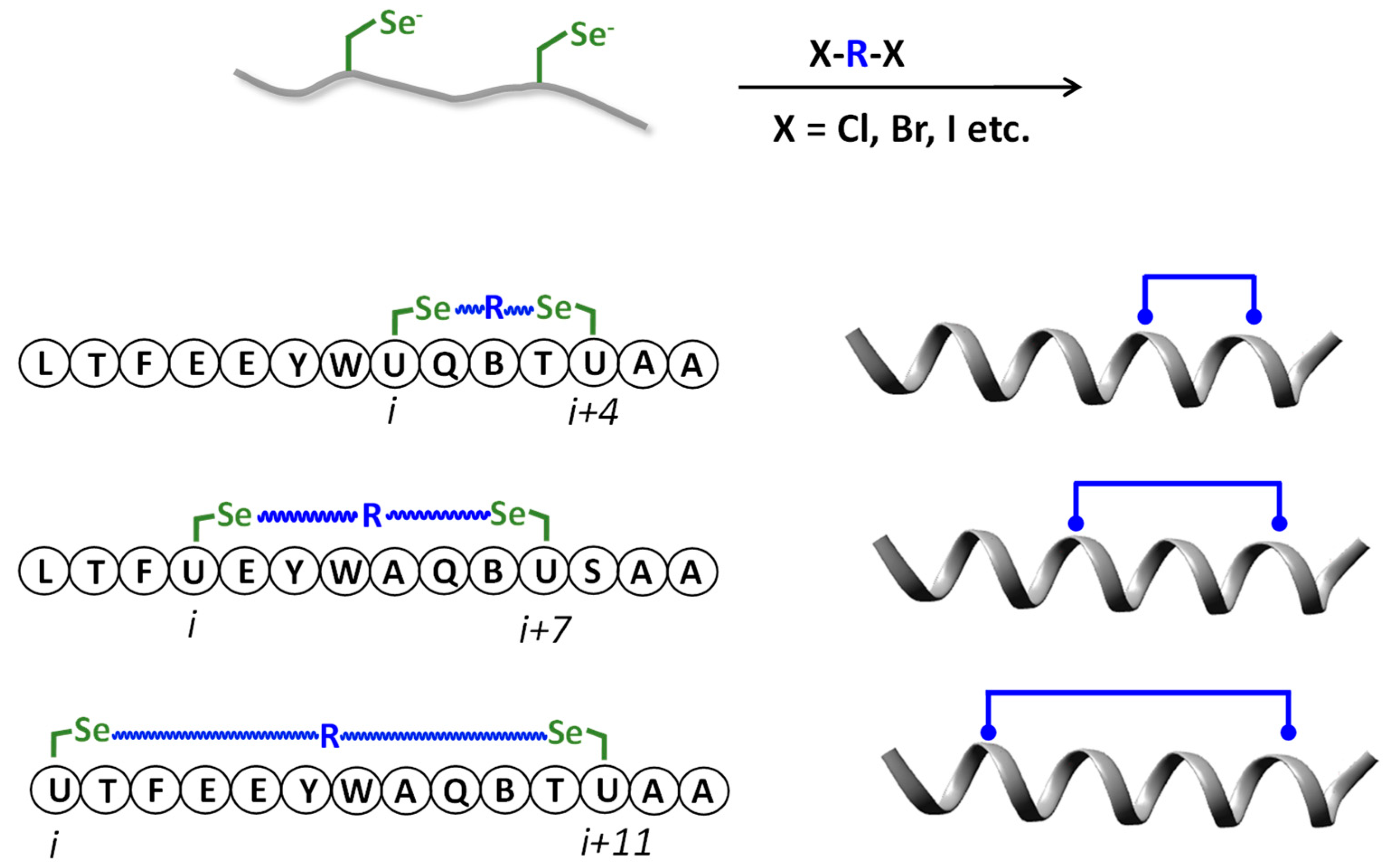
6. Selenopeptides Cyclization and Stapling
7. Modified Peptides Containing Selenium
8. Derivatization Based on Oxidation/Elimination: Conversion of Selenocysteine to Dehydroalanine
9. Photochemical Reactions of Selenium-Containing Peptides
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. The deiodinase family: Selenoenzymes regulating thyroid hormone availability and action. Cell. Mol. Life Sci. 2000, 57, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular Pathways and Physiological Roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; He, H.; Xiang, J.; Yin, H.; Hou, T. Selenium-Containing Proteins/Peptides from Plants: A Review on the Structures and Functions. J. Agric. Food Chem. 2020, 68, 15061–15073. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Roh, Y.J.; Han, S.J.; Park, I.; Lee, H.M.; Ok, Y.S.; Lee, B.C.; Lee, S.R. Role of Selenoproteins in Redox Regulation of Signaling and the Antioxidant System: A Review. Antioxidants 2020, 9, 383. [Google Scholar] [CrossRef]
- Mita, Y.; Nakayama, K.; Inari, S.; Nishito, Y.; Yoshioka, Y.; Sakai, N.; Sotani, K.; Nagamura, T.; Kuzuhara, Y.; Inagaki, K.; et al. Selenoprotein P-neutralizing antibodies improve insulin secretion and glucose sensitivity in type 2 diabetes mouse models. Nat. Commun. 2017, 8, 1658. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, L.; Orho-Melander, M.; Struck, J.; Bergmann, A.; Melander, O. Selenoprotein-P Deficiency Predicts Cardiovascular Disease and Death. Nutrients 2019, 11, 1852. [Google Scholar] [CrossRef] [Green Version]
- Reich, H.J.; Hondal, R.J. Why Nature Chose Selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- White, P.J. Selenium metabolism in plants. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2333–2342. [Google Scholar] [CrossRef]
- Walczak, R.; Westhof, E.; Carbon, P.; Krol, A. A novel RNA structural motif in the selenocysteine insertion element of eukaryotic selenoprotein mRNAs. RNA 1996, 2, 367–379. [Google Scholar] [PubMed]
- Chung, C.Z.; Krahn, N. The selenocysteine toolbox: A guide to studying the 21st amino acid. Arch. Biochem. Biophys. 2022, 730, 109421. [Google Scholar] [CrossRef] [PubMed]
- Muttenthaler, M.; Alewood, P.F. Selenopeptide chemistry. J. Pept. Sci. 2008, 14, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Shen, N.; Zhang, J.; Zhu, J.; Guo, Y.; Xu, L. A novel nanozyme based on selenopeptide-modified gold nanoparticles with a tunable glutathione peroxidase activity. RSC Adv. 2020, 10, 8685–8691. [Google Scholar] [CrossRef] [PubMed]
- Gokula, R.P.; Mahato, J.; Tripathi, A.; Singh, H.B.; Chowdhury, A. Self-Assembly of Nicotinic Acid-Conjugated Selenopeptides into Mesotubes. ACS Appl. Bio Mater. 2021, 4, 1912–1919. [Google Scholar] [CrossRef]
- Ahmed, K.; Ashraf, D.; Chotana, G.A.; Faisal, A.; Khan, K.M.; Salem, R.S.Z. Selenium-containing Peptides and their Biological Applications. Curr. Med. Chem. 2022, 29, 6379–6421. [Google Scholar] [CrossRef]
- Moroder, L. Isosteric replacement of sulfur with other chalcogens in peptides and proteins. J. Pept. Sci. 2005, 11, 187–214. [Google Scholar] [CrossRef]
- Pleasants, J.C.; Guo, W.; Rabenstein, D.L. A comparative study of the kinetics of selenol/diselenide and thiol/disulfide exchange reactions. J. Am. Chem. Soc. 1989, 111, 6553–6558. [Google Scholar] [CrossRef]
- Steinmann, D.; Nauser, T.; Koppenol, W.H. Selenium and Sulfur in Exchange Reactions: A Comparative Study. J. Org. Chem. 2010, 75, 6696–6699. [Google Scholar] [CrossRef]
- Koide, T.; Itoh, H.; Otaka, A.; Yasui, H.; Kuroda, M.; Esaki, N.; Soda, K.; Fujii, N. Synthetic study on selenocystine-containing peptides. Chem. Pharm. Bull. 1993, 41, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Arnold, A.P.; Tan, K.S.; Rabenstein, D.L. Nuclear magnetic resonance studies of the solution chemistry of metal complexes. 23. Complexation of methylmercury by selenohydryl-containing amino acids and related molecules. Inorg. Chem. 1986, 25, 2433–2437. [Google Scholar] [CrossRef]
- Huber, R.E.; Criddle, R.S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Connett, P.H.; Wetterhahn, K.E. Reaction of chromium (VI) with thiols: pH dependence of the chromium (VI) thio ester formation. J. Am. Chem. Soc. 1986, 108, 1842–1847. [Google Scholar] [CrossRef]
- Tajc, S.G.; Tolbert, B.S.; Basavappa, R.; Miller, B.L. Direct Determination of Thiol pKa by Isothermal Titration Microcalorimetry. J. Am. Chem. Soc. 2004, 126, 10508–10509. [Google Scholar] [CrossRef]
- Besse, D.; Siedler, F.; Diercks, T.; Kessler, H.; Moroder, L. The Redox Potential of Selenocystine in Unconstrained Cyclic Peptides. Angew. Chem. Int. Ed. Engl. 1997, 36, 883–885. [Google Scholar] [CrossRef]
- Lyles, M.M.; Gilbert, H.F. Catalysis of the Oxidative Folding of Ribonuclease A by Protein Disulfide Isomerase: Dependence of the Rate on the Composition of the Redox Buffer. Biochemistry 1991, 30, 613–619. [Google Scholar] [CrossRef]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfidebond stability. Methods Enzymol. 1995, 251, 8–28. [Google Scholar] [CrossRef]
- Beld, J.; Woycechowsky, K.J.; Hilvert, D. Selenoglutathione: Efficient Oxidative Protein Folding by a Diselenide. Biochemistry 2007, 46, 5382–5390. [Google Scholar] [CrossRef] [PubMed]
- Kildahl, N.K. Bond Energy Data Summarized. J. Chem. Educ. 1995, 72, 423–424. [Google Scholar] [CrossRef]
- Nauser, T.; Steinmann, D.; Grassi, G.; Koppenol, W.H. Why Selenocysteine Replaces Cysteine in Thioredoxin Reductase: A Radical Hypothesis. Biochemistry 2014, 53, 5017–5022. [Google Scholar] [CrossRef]
- Fan, F.; Ji, S.; Sun, C.; Liu, C.; Yu, Y.; Fu, Y.; Xu, H. Wavelength-Controlled Dynamic Metathesis: A Light-Driven Exchange Reaction between Disulfide and Diselenide Bonds. Angew. Chem. Int. Ed. 2018, 57, 16426–16430. [Google Scholar] [CrossRef]
- Merrifield, R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar] [CrossRef]
- Dawson, P.E.; Muir, T.W.; Clark-Lewis, I.; Kent, S.B.H. Synthesis of proteins by native chemical ligation. Science 1994, 266, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Torbeev, V.Y.; Kent, S.B.H. Convergent Chemical Synthesis and Crystal Structure of a 203 Amino Acid “Covalent Dimer” HIV-1 Protease Enzyme Molecule. Angew. Chem. Int. Ed. 2007, 46, 1667–1670. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, I.; Tezuka, K.; Fukae, K.; Ishii, K.; Taduru, K.; Maeda, M.; Ouchi, M.; Yoshida, K.; Nambu, Y.; Igarashi, J.; et al. Chemical Synthesis of Homogeneous Human Glycosyl-Interferon-β That Exhibits Potent Antitumor Activity in Vivo. J. Am. Chem. Soc. 2012, 134, 5428–5431. [Google Scholar] [CrossRef]
- Bang, D.; Kent, S.B.H. A One-Pot Total Synthesis of Crambin. Angew. Chem. Int. Ed. 2004, 43, 2534–2538. [Google Scholar] [CrossRef]
- Wintermann, F.; Engelbrecht, S. Reconstitution of the Catalytic Core of F-ATPase (Aβ)3γ from Escherichia Coli Using Chemically Synthesized Subunit γ. Angew. Chem. Int. Ed. 2013, 52, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Bondalapati, S.; Jbara, M.; Brik, A. Expanding the Chemical Toolbox for the Synthesis of Large and Uniquely Modified Proteins. Nat. Chem. 2016, 8, 407–418. [Google Scholar] [CrossRef]
- Hackeng, T.M.; Griffin, J.H.; Dawson, P.E. Protein Synthesis by Native Chemical Ligation: Expanded Scope by Using Straightforward Methodology. Proc. Natl. Acad. Sci. USA 1999, 96, 10068–10073. [Google Scholar] [CrossRef] [Green Version]
- Gieselman, M.D.; Xie, L.; van der Donk, W.A. Synthesis of a Selenocysteine-Containing Peptide by Native Chemical Ligation. Org. Lett. 2001, 3, 1331–1334. [Google Scholar] [CrossRef]
- Quaderer, R.; Sewing, A.; Hilvert, D. Selenocysteine-mediated native chemical ligation. Helv. Chim. Acta 2001, 84, 1197–1206. [Google Scholar]
- Hondal, R.J.; Nilsson, B.L.; Raines, R.T. Selenocysteine in Native Chemical Ligation and Expressed Protein Ligation. J. Am. Chem. Soc. 2001, 123, 5140–5141. [Google Scholar] [CrossRef]
- Nauser, T.; Dockheer, S.; Kissner, R.; Koppenol, W.H. Catalysis of Electron Transfer by Selenocysteine. Biochemistry 2006, 45, 6038–6043. [Google Scholar] [CrossRef]
- Guenther, W.H.H. Methods in selenium chemistry. III. Reduction of diselenides with dithiothreitol. J. Org. Chem. 1967, 32, 3931–3933. [Google Scholar] [CrossRef]
- Sayers, J.; Payne, R.J.; Winssinger, N. Peptide nucleic acid-templated selenocystine–selenoester ligation enables rapid miRNA detection. Chem. Sci. 2018, 9, 896–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chisholm, T.S.; Kulkarni, S.S.; Hossain, K.R.; Cornelius, F.; Clarke, R.J.; Payne, R.J. Peptide Ligation at High Dilution via Reductive Diselenide-Selenoester Ligation. J. Am. Chem. Soc. 2020, 142, 1090–1100. [Google Scholar] [CrossRef]
- Mitchell, N.J.; Malins, L.R.; Liu, X.; Thompson, R.E.; Chan, B.; Radom, L.; Payne, R.J. Rapid Additive-Free Selenocystine–Selenoester Peptide Ligation. J. Am. Chem. Soc. 2015, 137, 14011–14014. [Google Scholar] [CrossRef]
- Agouridas, V.; El Mahdi, O.; Diemer, V.; Cargoët, M.; Monbaliu, J.C.M.; Melnyk, O. Native Chemical Ligation and Extended Methods: Mechanisms, Catalysis, Scope, and Limitations. Chem. Rev. 2019, 119, 7328–7443. [Google Scholar] [CrossRef]
- Jakubke, H.D. Über die Verwendung von Aminosäure-und Peptidverbindungen des Selenophenols als “aktivierte Ester” zur Peptidsynthese. Z. Chem. 1963, 3, 65–66. [Google Scholar] [CrossRef]
- Hanna, C.C.; Kulkarni, S.S.; Watson, E.E.; Premdjee, B.; Payne, R.J. Solid-phase synthesis of peptide selenoesters via a side-chain anchoring strategy. Chem. Commun. 2017, 53, 5424–5427. [Google Scholar] [CrossRef] [Green Version]
- Ghassemian, A.; Vila-Farrés, X.; Alewood, P.F.; Durek, T. Solid phase synthesis of peptide-selenoesters. Bioorg. Med. Chem. 2013, 21, 3473–3478. [Google Scholar] [CrossRef] [PubMed]
- Hackenberger, C.P.R.; Schwarzer, D. Chemoselective Ligation and Modification Strategies for Peptides and Proteins. Angew. Chem. Int. Ed. 2008, 47, 10030–10074. [Google Scholar] [CrossRef] [PubMed]
- Wan, Q.; Danishefsky, S.J. Free-Radical-Based, Specific Desulfurization of Cysteine: A Powerful Advance in the Synthesis of Polypeptides and Glycopolypeptides. Angew. Chem. Int. Ed. 2007, 46, 9248–9252. [Google Scholar] [CrossRef]
- Chiang, K.P.; Jensen, M.S.; McGinty, R.K.; Muir, T.W. A Semisynthetic Strategy to Generate Phosphorylated and Acetylated Histone H2B. ChemBioChem 2009, 10, 2182–2187. [Google Scholar] [CrossRef] [PubMed]
- Metanis, N.; Keinan, E.; Dawson, P.E. Traceless Ligation of Cysteine Peptides Using Selective Deselenization. Angew. Chem. Int. Ed. 2010, 49, 7049–7053. [Google Scholar] [CrossRef] [Green Version]
- Dery, S.; Reddy, P.S.; Dery, L.; Mousa, R.; Dardashti, R.N.; Metanis, N. Insights into the Deselenization of Selenocysteine into Alanine and Serine. Chem. Sci. 2015, 6, 6207–6212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malins, L.R.; Mitchell, N.J.; McGowan, S.; Payne, R.J. Oxidative Deselenization of Selenocysteine: Applications for Programmed Ligation at Serine. Angew. Chem. 2015, 127, 12907–12912. [Google Scholar] [CrossRef]
- Reddy, P.S.; Dery, S.; Metanis, N. Chemical Synthesis of Proteins with Non-Strategically Placed Cysteines Using Selenazolidine and Selective Deselenization. Angew. Chem. 2016, 128, 1004–1007. [Google Scholar] [CrossRef]
- Dery, L.; Reddy, P.S.; Dery, S.; Mousa, R.; Ktorza, O.; Talhami, A.; Metanis, N. Accessing human selenoproteins through chemical protein synthesis. Chem. Sci. 2017, 8, 1922–1926. [Google Scholar] [CrossRef] [Green Version]
- Ollivier, N.; Blanpain, A.; Boll, E.; Raibaut, L.; Drobecq, H.; Melnyk, O. Selenopeptide Transamidation and Metathesis. Org. Lett. 2014, 16, 4032–4035. [Google Scholar] [CrossRef]
- Thornton, J.M. Disulphide bridges in globular proteins. J. Mol. Biol. 1981, 151, 261–287. [Google Scholar] [CrossRef]
- Creighton, T.E.; Zapun, A.; Darby, N.J. Mechanisms and catalysts of disulphide bond formation in proteins. Trends Biotechnol. 1995, 13, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Buchner, J.; Moroder, L. Oxidative Folding of Peptides and Proteins; RSC Publishing: Cambridge, UK, 2009; Volume 15. [Google Scholar]
- Pegoraro, S.; Fiori, S.; Cramer, J.; Rudolph-Böhner, S.; Moroder, L. The disulfide-coupled folding pathway of apamin as derived from diselenide-quenched analogs and intermediates. Protein Sci. 1999, 8, 1605–1613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pegoraro, S.; Fiori, S.; Rudolph-Böhner, S.; Watanabe, T.X.; Moroder, L. Isomorphous replacement of cystine with selenocystine in endothelin: Oxidative refolding, biological and conformational properties of [Sec3,Sec11,Nle7]-endothelin-1. J. Mol. Biol. 1998, 284, 779–792. [Google Scholar] [CrossRef]
- Arai, K.; Mikami, R. Redox Chemistry of Selenols and Diselenides as Potential Manipulators for Structural Maturation of Peptides and Proteins. Met. Res. 2022, 2, rev-1–rev-17. [Google Scholar] [CrossRef]
- Walewska, A.; Zhang, M.M.; Skalicky, J.J.; Yoshikami, D.; Olivera, B.M.; Bulaj, G. Integrated Oxidative Folding of Cysteine/Selenocysteine Containing Peptides: Improving Chemical Synthesis of Conotoxins. Angew. Chem. Int. Ed. 2009, 48, 2221–2224. [Google Scholar] [CrossRef] [PubMed]
- Weil-Ktorza, O.; Rege, N.; Lansky, S.; Shalev, D.E.; Shoham, G.; Weiss, M.A.; Metanis, N. Substitution of an Internal Disulfide Bridge with a Diselenide Enhances both Foldability and Stability of Human Insulin. Chem. Eur. J. 2019, 25, 8513–8521. [Google Scholar] [CrossRef]
- Medini, K.; Harris, P.W.R.; Menorca, A.; Hards, K.; Cook, G.M.; Brimble, M.A. Synthesis and activity of a diselenide bond mimetic of the antimicrobial protein caenopore-5. Chem. Sci. 2016, 7, 2005–2010. [Google Scholar] [CrossRef] [Green Version]
- Takei, T.; Tanaka, H.; Okumura, N.; Takao, T.; Moroder, L.; Hojo, H. Chemical synthesis of per-selenocysteine human epidermal growth factor. J. Pept. Sci. 2022. [Google Scholar] [CrossRef]
- Muttenthaler, M.; Nevin, S.T.; Grishin, A.A.; Ngo, S.T.; Choy, P.T.; Daly, N.L.; Hu, S.H.; Armishaw, C.J.; Wang, C.I.A.; Lewis, R.J.; et al. Solving the α-Conotoxin Folding Problem: Efficient Selenium-Directed On-Resin Generation of More Potent and Stable Nicotinic Acetylcholine Receptor Antagonists. J. Am. Chem. Soc. 2010, 132, 3514–3522. [Google Scholar] [CrossRef]
- Arai, K.; Takei, T.; Okumura, M.; Watanabe, S.; Amagai, Y.; Asahina, Y.; Moroder, L.; Hojo, H.; Inaba, K.; Iwaoka, M. Preparation of Selenoinsulin as a Long-Lasting Insulin Analogue. Angew. Chem. Int. Ed. 2017, 56, 5522–5526. [Google Scholar] [CrossRef]
- Armishaw, C.J.; Daly, N.L.; Nevin, S.T.; Adams, D.J.; Craik, D.J.; Alewood, P.F. α-Selenoconotoxins, a New Class of Potent α7 Neuronal Nicotinic Receptor Antagonists. J. Biol. Chem. 2006, 281, 14136–14143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Araujo, A.D.; Callaghan, B.; Nevin, S.T.; Daly, N.L.; Craik, D.J.; Moretta, M.; Hopping, G.; Christie, M.J.; Adams, D.J.; Alewood, P.F. Total Synthesis of the Analgesic Conotoxin MrVIB through Selenocysteine-Assisted Folding. Angew. Chem. Int. Ed. 2011, 50, 6527–6529. [Google Scholar] [CrossRef] [PubMed]
- Walewska, A.; Jaśkiewicz, A.; Bulaj, G.; Rolka, K. Selenopeptide Analogs of EETI-II Retain Potent Trypsin Inhibitory Activities. Chem. Biol. Drug Des. 2011, 77, 93–97. [Google Scholar] [CrossRef]
- Gowd, K.H.; Yarotskyy, V.; Elmslie, K.S.; Skalicky, J.J.; Olivera, B.M.; Bulaj, G. Site-Specific Effects of Diselenide Bridges on the Oxidative Folding of a Cystine Knot Peptide, ω-Selenoconotoxin GVIA. Biochemistry 2010, 49, 2741–2752. [Google Scholar] [CrossRef] [Green Version]
- Steiner, A.M.; Woycechowsky, K.J.; Olivera, B.M.; Bulaj, G. Reagentless Oxidative Folding of Disulfide-Rich Peptides Catalyzed by an Intramolecular Diselenide. Angew. Chem. Int. Ed. 2012, 51, 5580–5584. [Google Scholar] [CrossRef] [Green Version]
- Mousa, R.; Hidmi, T.; Pomyalov, S.; Lansky, S.; Khouri, L.; Shalev, D.E.; Shoham, G.; Metanis, N. Diselenide crosslinks for enhanced and simplified oxidative protein folding. Commun. Chem. 2021, 4, 30. [Google Scholar] [CrossRef]
- Metanis, N.; Hilvert, D. Strategic Use of Non-Native Diselenide Bridges to Steer Oxidative Protein Folding. Angew. Chem. Int. Ed. 2012, 51, 5585–5588. [Google Scholar] [CrossRef]
- Metanis, N.; Hilvert, D. Harnessing selenocysteine reactivity for oxidative protein folding. Chem. Sci. 2015, 6, 322–325. [Google Scholar] [CrossRef] [Green Version]
- Beld, J.; Woycechowsky, K.J.; Hilvert, D. Catalysis of Oxidative Protein Folding by Small-Molecule Diselenides. Biochemistry 2008, 47, 6985–6987. [Google Scholar] [CrossRef]
- Reddy, P.S.; Metanis, N. Small molecule diselenide additives for in vitro oxidative protein folding. Chem. Commun. 2016, 52, 3336–3339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendil, D.; Demirci, Z.; Uluozlu, O.D.; Tuzen, M.; Soylak, M. A new separation and preconcentration method for selenium in some foods using modified silica gel with 2, 6-diamino-4-phenil-1, 3, 5- triazine. Food Chem. 2017, 221, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Uden, P.C.; Boakye, H.T.; Kahakachchi, C.; Tyson, J.F. Selective detection and identification of Se containing compounds– review and recent developments. J. Chromatogr. A 2004, 1050, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Xu, Q.; Lee, C.; Zhang, H. Selenium speciation analysis of Misgurnus anguillicaudatus selenoprotein by HPLC− ICP−MS and HPLC−ESI−MS/MS. Eur. Food Res. Technol. 2012, 235, 169–176. [Google Scholar] [CrossRef]
- Liu, K.; Du, R.; Chen, F. Antioxidant activities of Se-MPS: A selenopeptide identified from selenized brown rice protein hydrolysates. LWT—Food Sci. Technol. 2019, 111, 555–560. [Google Scholar] [CrossRef]
- Giusti, P.; Schaumlöffel, D.; Encinar, J.R.; Szpunar, J. Interfacing reversed-phase nanoHPLC with ICP-MS and on-line isotope dilution analysis for the accurate quantification of selenium-containing peptides in protein tryptic digests. J. Anal. At. Spectrom. 2005, 20, 1101–1107. [Google Scholar] [CrossRef]
- Guo, L.; Yang, W.; Huang, Q.; Qiang, J.; Hart, J.R.; Wang, W.; Hu, J.; Zhu, J.; Liu, N.; Zhang, Y. Selenocysteine-Specific Mass Spectrometry Reveals Tissue-Distinct Selenoproteomes and Candidate Selenoproteins. Cell Chem. Biol. 2018, 25, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Bak, D.W.; Gao, J.; Wang, C.; Weerapana, E. A Quantitative Chemoproteomic Platform to Monitor Selenocysteine Reactivity within a Complex Proteome. Cell Chem. Biol. 2018, 25, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Waliczek, M.; Pehlivan, Ö.; Stefanowicz, P. A photochemical transformation of cyclic peptides leading to formation of selenolanthionine bridges. New J. Chem. 2020, 44, 11433–11436. [Google Scholar] [CrossRef]
- de Araujo, A.D.; Mobli, M.; King, G.F.; Alewood, P.F. Cyclization of Peptides by using Selenolanthionine Bridges. Angew. Chem. Int. Ed. 2012, 51, 10298–10302. [Google Scholar] [CrossRef]
- de Araujo, A.D.; Perry, S.R.; Fairlie, D.P. Chemically Diverse Helix-Constrained Peptides Using Selenocysteine Crosslinking. Org. Lett. 2018, 20, 1453–1456. [Google Scholar] [CrossRef]
- Rew, Y.; Malkmus, S.; Svensson, C.; Yaksh, T.L.; Chung, N.N.; Schiller, P.W.; Cassel, J.A.; DeHaven, R.N.; Goodman, M. Synthesis and Biological Activities of Cyclic Lanthionine Enkephalin Analogues: δ-Opioid Receptor Selective Ligands. J. Med. Chem. 2002, 45, 3746–3754. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, C.; Paul, M.; Xie, L.; van der Donk, W.A. Biosynthesis and Mode of Action of Lantibiotics. Chem. Rev. 2005, 105, 633–684. [Google Scholar] [CrossRef]
- Barreto, A.F.S.; Vercillo, O.E.; Birkett, M.A.; Caulfield, J.C.; Wessjohann, L.A.; Andrade, C.K.Z. Fast and efficient microwave-assisted synthesis of functionalized peptoids via Ugi reactions. Org. Biomol. Chem. 2011, 9, 5024–5027. [Google Scholar] [CrossRef] [PubMed]
- Ridder, B.; Mattes, D.S.; Nesterov-Mueller, A.; Breitling, F.; Meier, M.A.R. Peptide array functionalization via the Ugi four-component reaction. Chem. Commun. 2017, 53, 5553–5556. [Google Scholar] [CrossRef]
- Hartweg, M.; Edwards-Gayle, C.J.C.; Radvar, E.; Collis, D.; Reza, M.; Kaupp, M.; Steinkoenig, J.; Ruokolainen, J.; Rambo, R.; Barner-Kowollik, C.; et al. Ugi multicomponent reaction to prepare peptide–peptoid hybrid structures with diverse chemical functionalities. Polym. Chem. 2018, 9, 482–489. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Wang, S.; Liu, J.; Xie, Z.; Luan, S.; Xiao, C.; Tao, Y.; Wang, X. Ugi Reaction of Natural Amino Acids: A General Route toward Facile Synthesis of Polypeptoids for Bioapplications. ACS Macro Lett. 2016, 5, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Ugi, I.; Steinbrückner, C. Isonitrile. II. Reaktion von Isonitrilen mit Carbonylverbindungen, Aminen und Stickstoffwasserstoffsäure. Chem. Ber. 1961, 94, 734–742. [Google Scholar] [CrossRef]
- Abbas, M.; Bethke, J.; Wessjohann, L.A. One pot synthesis of selenocysteine containing peptoid libraries by Ugi multicomponent reactions in water. Chem. Commun. 2006, 541–543. [Google Scholar] [CrossRef]
- Yudin, A.K. Aziridines and Epoxides in Organic Synthesis, 1st ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006. [Google Scholar]
- Kowalczyk, A.; Pieczonka, A.M.; Rachwalski, M.; Leśniak, S.; Stączek, P. Synthesis and Evaluation of Biological Activities of Aziridine Derivatives of Urea and Thiourea. Molecules 2018, 23, 45. [Google Scholar] [CrossRef] [Green Version]
- Fürmeier, S.; Metzger, J.O. Fat-Derived Aziridines and Their N-Substituted Derivatives: Biologically Active Compounds Based on Renewable Raw Materials. Eur. J. Org. Chem. 2003, 649–659. [Google Scholar] [CrossRef]
- Bae, J.H.; Shin, S.H.; Park, C.S.; Lee, W.K. Preparation of Cysteinol Derivatives by Highly Regioselective Ring Opening of Nonactivated Chiral Aziridines by Thiols. Tetrahedron 1999, 55, 10041–10046. [Google Scholar] [CrossRef]
- Galonić, D.P.; van der Donk, W.A.; Gin, D.Y. Site-Selective Conjugation of Thiols with Aziridine-2-CarboxylicAcid-Containing Peptides. J. Am. Chem. Soc. 2004, 126, 12712–12713. [Google Scholar] [CrossRef]
- Spork, A.P.; Donohoe, T.J. Aziridine electrophiles in the functionalisation of peptide chains with amine nucleophiles. Org. Biomol. Chem. 2015, 13, 8545–8549. [Google Scholar] [CrossRef]
- Braga, A.L.; Lüdtke, D.S.; Paixão, M.W.; Alberto, E.E.; Stefani, H.A.; Juliano, L. Straightforward Synthesis of Non-Natural Selenium Containing Amino Acid Derivatives and Peptides. Eur. J. Org. Chem. 2005, 4260–4264. [Google Scholar] [CrossRef]
- Braga, A.L.; Paixão, M.W.; Lüdtke, D.S.; Silveira, C.C.; Rodrigues, O.E.D. Synthesis of New Chiral Aliphatic Amino Diselenides and Their Application as Catalysts for the Enantioselective Addition of Diethylzinc to Aldehydes. Org. Lett. 2003, 5, 2635–2638. [Google Scholar] [CrossRef] [PubMed]
- Arsenyan, P.; Lapcinska, S.; Ivanova, A.; Vasiljeva, J. Peptide Functionalization Through the Generation of Selenocysteine Electrophile. Eur. J. Org. Chem. 2019, 4951–4961. [Google Scholar] [CrossRef]
- Baldwin, J.E. Rules for Ring Closure. J. Chem. Soc. Chem. Commun. 1976, 734–736. [Google Scholar] [CrossRef]
- Sajid, M.A.; Khan, Z.A.; Shahzad, S.A.; Naqvi, S.A.R.; Usman, M. 5-endo-dig cyclizations in organic syntheses. Mol. Divers. 2020, 24, 295–317. [Google Scholar] [CrossRef]
- Lapcinska, S.; Arsenyan, P. Selenocystine Peptides Performance in 5-endo-dig Reactions. Eur. J. Org. Chem. 2020, 784–795. [Google Scholar] [CrossRef]
- Lapcinska, S.; Arsenyan, P. Selenocysteinyl electrophiles efficiently promote the formation of coumarin and quinolinone cores by 6-endo-dig cyclization. New J. Chem. 2021, 45, 16625–16634. [Google Scholar] [CrossRef]
- Gross, E.; Morell, J.L. The presence of dehydroalanine in the antibiotic nisin and its relationship to activity. J. Am. Chem. Soc. 1967, 89, 2791–2792. [Google Scholar] [CrossRef] [PubMed]
- Ebata, M.; Miyazaki, K.; Otsuka, H. Studies on Siomycin. II the Composition and Degradation Products of Siomycin A. J. Antibiot. 1969, 22, 423–433. [Google Scholar] [CrossRef] [Green Version]
- Bodanszky, M.; Scozzie, J.A.; Muramatsu, I. Dehydroalanine Residues in Thiostrepton. J. Antibiot. 1970, 23, 9–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, R.; Roy, J. Selenomethionine, a Potential Catalytic Antioxidant in Biological Systems. J. Org. Chem. 1971, 36, 2561–2563. [Google Scholar] [CrossRef]
- Okeley, N.M.; Zhu, Y.; van der Donk, W.A. Facile Chemoselective Synthesis of Dehydroalanine-Containing Peptides. Org. Lett. 2000, 2, 3603–3606. [Google Scholar] [CrossRef]
- Hashimoto, K.; Sakai, M.; Okuno, T.; Shirahama, H. β-Phenylselenoalanine as a dehydroalanine precursor-efficient synthesis of alternariolide (AM-toxin I). Chem. Commun. 1996, 1139–1140. [Google Scholar] [CrossRef]
- Ma, S.; Caprioli, R.M.; Hill, K.E.; Burk, R.F. Loss of selenium from selenoproteins: Conversion of selenocysteine to dehydroalanine in vitro. J. Am. Soc. Mass. Spectrom. 2003, 14, 593–600. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.K.; Weaver, J.D.; Zhang, S.; Lei, X.G. Knockout of SOD1 promotes conversion of selenocysteine to dehydroalanine in murine hepatic GPX1 protein. Free Radic. Biol. Med. 2011, 51, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Dadová, J.; Galan, S.R.G.; Davis, B.G. Synthesis of modified proteins via functionalization of dehydroalanine. Curr. Opin. Chem. Biol. 2018, 46, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lyons, B.; Truscott, R.J.W.; Schey, K.L. Human protein aging: Modification and crosslinking through dehydroalanine and dehydrobutyrine intermediates. Aging Cell 2014, 13, 226–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.; Tao, H.; Szymczak, L.C.; Lin, L.; Song, J.; Wang, Y.; Bai, S.; Modica, J.; Huang, S.Y.; Mrksich, M.; et al. Efficient Enzymatic Incorporation of Dehydroalanine Based on SAMDI-Assisted Identification of Optimized Tags for OspF/SpvC. ACS Chem. Biol. 2022, 17, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Chen, H.; Mu, N.; Wang, W.; Zhu, K.; Ruan, Z.; Wang, S. Nosiheptide analogues as potential antibacterial agents via dehydroalanine region modifications: Semi-synthesis, antimicrobial activity and molecular docking study. Bioorganic Med. Chem. 2021, 31, 115970. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Dynamic Diselenide Bonds: Exchange Reaction Induced by Visible Light without Catalysis. Angew. Chem. Int. Ed. 2014, 53, 6781–6785. [Google Scholar] [CrossRef]
- Kang, X.; Yuan, Y.; Xu, H.; Chen, Y. Thermal- and Light-driven Metathesis Reactions Between Different Diselenides. Chem. Res. Chin. Univ. 2022, 38, 516–521. [Google Scholar] [CrossRef]
- Suzuki, N.; Takahashi, A.; Ohishi, T.; Goseki, R.; Otsuka, H. Enhancement of the stimuli-responsiveness and photo-stability of dynamic diselenide bonds and diselenide-containing polymers by neighboring aromatic groups. Polymer 2018, 154, 281–290. [Google Scholar] [CrossRef]
- Waliczek, M.; Pehlivan, Ö.; Stefanowicz, P. Light-Driven Diselenide Metathesis in Peptides. ChemistryOpen 2019, 8, 1199–1203. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Xia, J.; Cao, M.; Xu, H. Wavelength-Controlled Light-Responsive Polymer Vesicle Based on Se−S Dynamic Chemistry. ACS Macro Lett. 2020, 9, 163–168. [Google Scholar] [CrossRef]
- Dowman, L.J.; Kulkarni, S.S.; Alegre-Requena, J.V.; Giltrap, A.M.; Norman, A.R.; Sharma, A.; Gallegos, L.C.; Mackay, A.S.; Welegedara, A.P.; Watson, E.E.; et al. Site-selective photocatalytic functionalization of peptides and proteins at selenocysteine. Nat. Commun. 2022, 13, 6885. [Google Scholar] [CrossRef]
- Lapcinska, S.; Dimitrijevs, P.; Lapcinskis, L.; Arsenyan, P. Visible Light-Mediated Functionalization of Selenocystine-Containing Peptides. Adv. Synth. Catal. 2021, 363, 3318–3328. [Google Scholar] [CrossRef]
- Lapcinska, S.; Dimitrijevs, P.; Arsenyan, P. Visible Light-Mediated Synthesis of Se−S Bond-Containing Peptides. Adv. Synth. Catal. 2021, 363, 3968–3972. [Google Scholar] [CrossRef]
- Cui, J.; del Campo, A. Multivalent H-bonds for self-healing hydrogels. Chem. Commun. 2012, 48, 9302–9304. [Google Scholar] [CrossRef] [PubMed]
- Nakahata, M.; Takashima, Y.; Yamaguchi, H.; Harada, A. Redox-responsive self-healing materials formed from host−guest polymers. Nat. Commun. 2011, 2, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Su, Q.; Zhang, J.; Dong, A.; Lin, C.; Zhang, J. Facile Access to Multisensitive and Self-Healing Hydrogels with Reversible and Dynamic Boronic Ester and Disulfide Linkages. Biomacromolecules 2017, 18, 1356–1364. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Xu, H.; Suzuki, N.; Takahashi, A.; Ohishi, T.; Goseki, R.; Xie, X.M.; Otsuka, H. Structural reorganization and crack-healing properties of hydrogels based on dynamic diselenide linkages. Sci. Technol. Adv. Mater. 2020, 21, 450–460. [Google Scholar] [CrossRef]
- An, X.; Aguirresarobe, R.H.; Irusta, L.; Ruipérez, F.; Matxain, J.M.; Pan, X.; Aramburu, N.; Mecerreyes, D.; Sardon, H.; Zhu, J. Aromatic diselenide crosslinkers to enhance the reprocessability and self-healing of polyurethane thermosets. Polym. Chem. 2017, 8, 3641–3646. [Google Scholar] [CrossRef]
- Ji, S.; Cao, W.; Yu, Y.; Xu, H. Visible-Light-Induced Self-Healing Diselenide-Containing Polyurethane Elastomer. Adv. Mater. 2015, 27, 7740–7745. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Deng, S.; Yan, T.; Zhang, X.; Tian, R.; Xu, J.; Sun, H.; Yu, S.; Liu, J. Biocompatible Diselenide-Containing Protein Hydrogels with Effective Visible-Light-Initiated Self-Healing Properties. Polymers 2021, 13, 4360. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pehlivan, Ö.; Waliczek, M.; Kijewska, M.; Stefanowicz, P. Selenium in Peptide Chemistry. Molecules 2023, 28, 3198. https://doi.org/10.3390/molecules28073198
Pehlivan Ö, Waliczek M, Kijewska M, Stefanowicz P. Selenium in Peptide Chemistry. Molecules. 2023; 28(7):3198. https://doi.org/10.3390/molecules28073198
Chicago/Turabian StylePehlivan, Özge, Mateusz Waliczek, Monika Kijewska, and Piotr Stefanowicz. 2023. "Selenium in Peptide Chemistry" Molecules 28, no. 7: 3198. https://doi.org/10.3390/molecules28073198