
Palladium-Catalyzed Stereoselective Construction of 1,3-Stereocenters Displaying Axial and Central Chirality via Asymmetric Alkylations
Abstract
:1. Introduction
2. Results and Discussion
2.1. Optimization of the Reaction Conditions
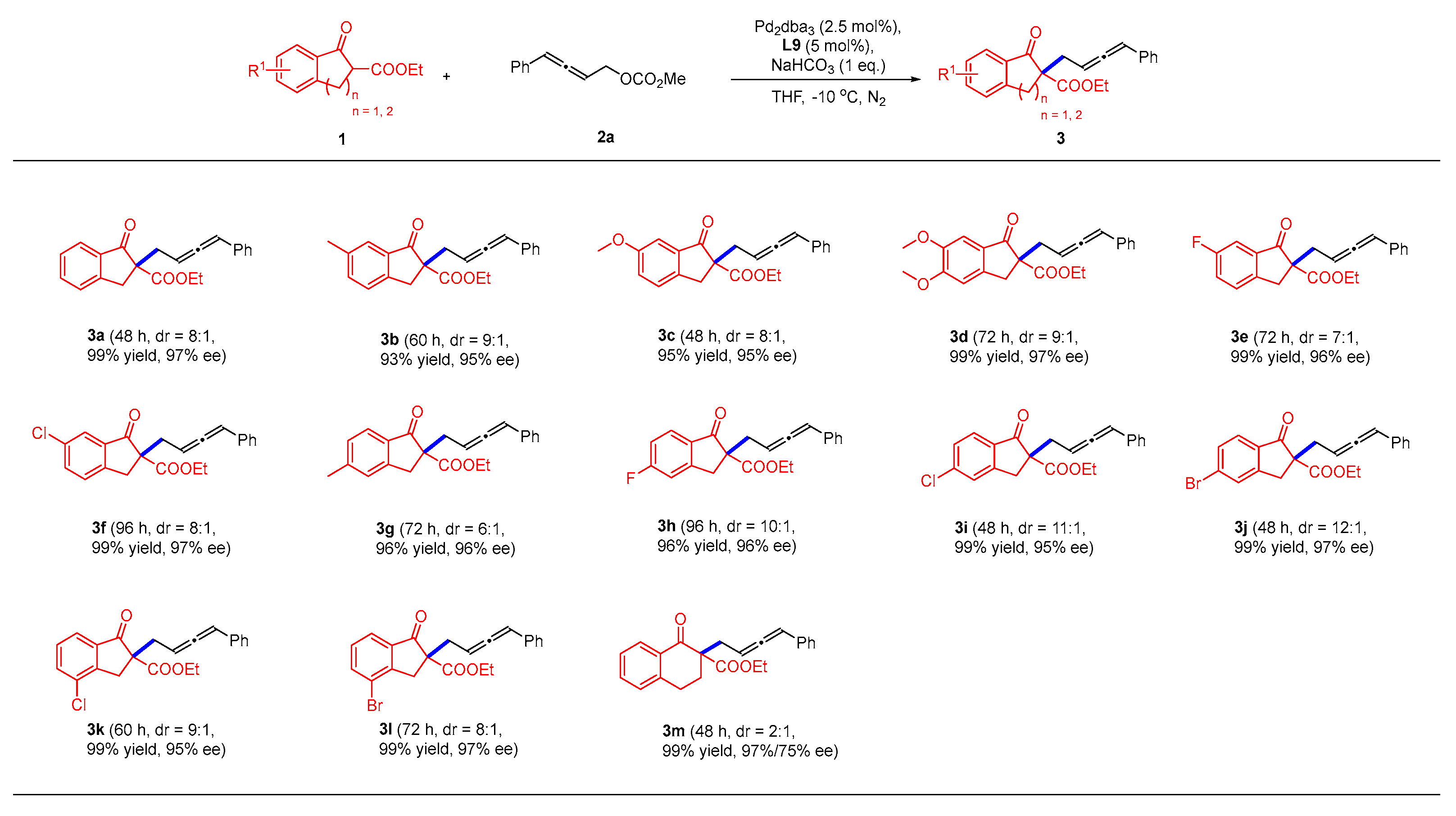
2.2. Substrate Scope for the Asymmetric Alkylations of β-Ketoester 1
2.3. Substrate Scope for the Asymmetric Alkylations of Allenylic Carbonates 2
2.4. Gram-Scale Reaction and Product Derivatization
2.5. Plausible Mechanism of the Palladium-Catalyzed Alkylation of β-Ketoester 1
3. Materials and Methods
3.1. General Information
3.2. Procedure for the Synthesis of Compounds 3
3.3. Procedure for the Synthesis of Compounds 4a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Liu, H.; Leow, D.; Huang, K.-W.; Tan, C.-H. Enantioselective Synthesis of Chiral Allenoates by Guanidine-Catalyzed Isomerization of 3-Alkynoates. J. Am. Chem. Soc. 2009, 131, 7212–7213. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, S.; Liu, N.; Werness, J.B.; Guzei, I.A.; Tang, W. Enantioselective Bromolactonization of Conjugated (Z)-Enynes. J. Am. Chem. Soc. 2010, 132, 3664–3665. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Z.; He, Z.; Li, G.; Hong, L.; Sun, W.; Wang, R. Organocatalytic Enantioselective Synthesis of Tetrasubstituted α-Amino Allenoates by Dearomative γ-Addition of 2,3-Disubstituted Indoles to β,γ-Alkynyl-α-imino Esters. Angew. Chem. Int. Ed. 2020, 59, 642–647. [Google Scholar] [CrossRef]
- Tap, A.; Blond, A.; Wakchaure, V.N.; List, B. Chiral Allenes via Alkynylogous Mukaiyama Aldol Reaction. Angew. Chem. Int. Ed. 2016, 55, 8962–8965. [Google Scholar] [CrossRef]
- Zheng, Y.; Miao, B.; Qin, A.; Xiao, J.; Liu, Q.; Li, G.; Zhang, L.; Zhang, F.; Guo, Y.; Ma, S. Negishi Coupling for Highly Selective Syntheses of Allenes via Ligand Effect and Mechanistic Study via SAESI-MS/MS. Chin. J. Chem. 2019, 37, 1003–1008. [Google Scholar] [CrossRef]
- Neff, R.K.; Frantz, D.E. Recent Advances in the Catalytic Syntheses of Allenes: A Critical Assessment. ACS Catal. 2014, 4, 519–528. [Google Scholar] [CrossRef]
- Ye, J.; Ma, S. Conquering three-carbon axial chirality of allenes. Org. Chem. Front. 2014, 1, 1210–1224. [Google Scholar] [CrossRef]
- Chu, W.-D.; Zhang, Y.; Wang, J. Recent advances in catalytic asymmetric synthesis of allenes. Catal. Sci. Technol. 2017, 7, 4570–4579. [Google Scholar] [CrossRef]
- Huang, X.; Ma, S. Allenation of Terminal Alkynes with Aldehydes and Ketones. Acc. Chem. Res. 2019, 52, 1301–1312. [Google Scholar] [CrossRef]
- Davis, C.R.; Luvaga, I.K.; Ready, J.M. Enantioselective Allylation of Alkenyl Boronates Promotes a 1,2-Metalate Rearrangement with 1,3-Diastereocontrol. J. Am. Chem. Soc. 2021, 143, 4921–4927. [Google Scholar] [CrossRef]
- Trost, B.M.; Schultz, J.E.; Chang, T.; Maduabum, M.R. Chemo-, Regio-, Diastereo-, and Enantioselective Palladium Allylic Alkylation of 1,3-Dioxaboroles as Synthetic Equivalents of α-Hydroxyketones. J. Am. Chem. Soc. 2019, 141, 9521–9526. [Google Scholar] [CrossRef]
- Trost, B.M.; Zell, D.; Hohn, C.; Mata, G.; Maruniak, A. Enantio-and Diastereoselective Synthesis of Chiral Allenes by Palladium-Catalyzed Asymmetric [3 + 2] Cycloaddition Reactions. Angew. Chem. Int. Ed. 2018, 130, 13098–13102. [Google Scholar] [CrossRef]
- Li, Z.; Hu, B.; Wu, Y.; Fei, C.; Deng, L. Control of chemoselectivity in asymmetric tandem reactions: Direct synthesis of chiral amines bearing nonadjacent stereocenters. Proc. Natl. Acad. Sci. USA 2018, 115, 1730–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, B.; Lee, R.; Li, J.; Ye, X.; Hong, S.N.; Qiu, S.; Coote, M.L.; Jiang, Z. Chemoselective Switch in the Asymmetric Organocatalysis of 5H-Oxazol-4-ones and N-Itaconimides: Addition–Protonation or [4 + 2] Cycloaddition. Angew. Chem. Int. Ed. 2016, 128, 1321–1325. [Google Scholar] [CrossRef]
- Suzuki, M.; Kurosawa, E. Okamurallene, a novel halogenated C15 metabolite from the red alga laurencia okamuhai yamada. Tetrahedron Lett. 1981, 22, 3853–3856. [Google Scholar] [CrossRef]
- Zhu, Y.L.; Pai, S.B.; Liu, S.H.; Grove, K.L.; Jones, B.C.; Simons, C.; Zemlicka, J.; Cheng, Y.C. Inhibition of replication of hepatitis B virus by cytallene in vitro. Antimicrob. Agents Chemother. 1997, 41, 1755–1760. [Google Scholar] [CrossRef] [Green Version]
- Cai, F.; Pu, X.; Qi, X.; Lynch, V.; Radha, A.; Ready, J.M. Chiral Allene-Containing Phosphines in Asymmetric Catalysis. J. Am. Chem. Soc. 2011, 133, 18066–18069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, X.; Qi, X.; Ready, J.M. Allenes in Asymmetric Catalysis: Asymmetric Ring Opening of meso-Epoxides Catalyzed by Allene-Containing Phosphine Oxides. J. Am. Chem. Soc. 2009, 131, 10364–10365. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Sone, Y.; Iwata, S.; Matsuzawa, H.; Mazaki, Y. Tetrathiafulvalenylallene: A New Class of Donor Molecules Having Strong Chiroptical Properties in Neutral and Doped States. Org. Lett. 2011, 13, 4688–4691. [Google Scholar] [CrossRef] [PubMed]
- Ching, C.K.; Lam, S.K. A comparison of two prostaglandin analogues (enprostil vs misoprostol) in the treatment of acute duodenal ulcer disease. J. Gastroenterol. 1995, 30, 607–614. [Google Scholar] [CrossRef]
- Watanabe, Y.; Yamazaki, T. Facile Preparation of CF3-Containing 1-Bromoallenes. Synlett 2009, 20, 3352–3354. [Google Scholar]
- Lü, B.; Jiang, X.; Fu, C.; Ma, S. Highly Regio- and Stereoselective Cyclic Iodoetherification of 4,5-Alkadienols. An Efficient Preparation of 2-(1′(Z)-Iodoalkenyl)tetrahydrofurans. J. Org. Chem. 2009, 74, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Yokota, M.; Fuchibe, K.; Ueda, M.; Mayumi, Y.; Ichikawa, J. Facile Synthesis of 1,1-Difluoroallenes via the Difluorovinylidenation of Aldehydes and Ketones. Org. Lett. 2009, 11, 3994–3997. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-Y.; Sun, X.-L.; Jing, Q.; Tang, Y. Enantioselective synthesis of allenic esters via an ylide route. Chem. Commun. 2006, 28, 2980–2982. [Google Scholar] [CrossRef]
- Skattebøfl, L. Chemistry of gem-dihalocyclopropanes—VI: A novel synthesis of cyclopentadienes and fulvenes. Tetrahedron 1967, 23, 1107–1117. [Google Scholar] [CrossRef]
- Woerly, E.M.; Cherney, A.H.; Davis, E.K.; Burke, M.D. Stereoretentive Suzuki−Miyaura Coupling of Haloallenes Enables Fully Stereocontrolled Access to (−)-Peridinin. J. Am. Chem. Soc. 2010, 132, 6941–6943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, G.; Xue, C.; Fu, C.; Ma, S. An Efficient Synthesis of Allenyl Perfluoroalkyl Ketones from Mono-1, 2-Addition-Elimination Reaction of Allenoates with RfMgX. Synlett 2010, 2, 281–285. [Google Scholar] [CrossRef]
- Deska, J.; del Pozo Ochoa, C.; Bäckvall, J.-E. Chemoenzymatic Dynamic Kinetic Resolution of Axially Chiral Allenes. Chem. Eur. J. 2010, 16, 4447–4451. [Google Scholar] [CrossRef]
- Qian, H.; Yu, X.; Zhang, J.; Sun, J. Organocatalytic Enantioselective Synthesis of 2,3-Allenoates by Intermolecular Addition of Nitroalkanes to Activated Enynes. J. Am. Chem. Soc. 2013, 135, 18020–18023. [Google Scholar] [CrossRef]
- Poulsen, P.H.; Li, Y.; Lauridsen, V.H.; Jørgensen, D.K.B.; Palazzo, T.A.; Meazza, M.; Jørgensen, K.A. Organocatalytic Formation of Chiral Trisubstituted Allenes and Chiral Furan Derivatives. Angew. Chem. Int. Ed. 2018, 57, 10661–10665. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Luo, H.; Zhang, Z.-M.; Zheng, W.-F.; Yin, Y.; Qian, H.; Zhang, J.; Ma, S. Pd-Catalyzed Enantioselective Syntheses of Trisubstituted Allenes via Coupling of Propargylic Benzoates with Organoboronic Acids. J. Am. Chem. Soc. 2020, 142, 9763–9771. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Greßies, S.; Chen, P.; Liu, G. Recent Advances and Perspectives in Transition Metal-Catalyzed 1,4-Functionalizations of Unactivated 1,3-Enynes for the Synthesis of Allenes. Chin. J. Chem. 2020, 38, 91–100. [Google Scholar] [CrossRef]
- Song, S.; Zhou, J.; Fu, C.; Ma, S. Catalytic enantioselective construction of axial chirality in 1,3-disubstituted allenes. Nat. Commun. 2019, 10, 507. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-C.; Hu, Y.-Z.; Wang, Z.-F.; Tao, H.-Y.; Wang, C.-J. Synergistic Cu/Pd-Catalyzed Asymmetric Allenylic Alkylation of Azomethine Ylides for the Construction of α-Allene-Substituted Nonproteinogenic α-Amino Acids. Chem. Eur. J. 2019, 25, 8681–8685. [Google Scholar] [CrossRef]
- Xiao, J.; Xu, H.; Huo, X.; Zhang, W.; Ma, S. One Stone Two Birds—Enantioselective Bimetallic Catalysis for α-Amino Acid Derivatives with an Allene Unit. Chin. J. Chem. 2021, 39, 1958–1964. [Google Scholar] [CrossRef]
- Wan, B.; Ma, S. Enantioselective decarboxylative amination: Synthesis of axially chiral allenyl amines. Angew. Chem. Int. Ed. 2013, 125, 459–463. [Google Scholar] [CrossRef]
- Zhu, C.; Chu, H.; Li, G.; Ma, S.; Zhang, J. Pd-Catalyzed Enantioselective Heck Reaction of Aryl Triflates and Alkynes. J. Am. Chem. Soc. 2019, 141, 19246–19251. [Google Scholar] [CrossRef]
- Chu, W.-D.; Zhang, L.; Zhang, Z.; Zhou, Q.; Mo, F.; Zhang, Y.; Wang, J. Enantioselective Synthesis of Trisubstituted Allenes via Cu(I)-Catalyzed Coupling of Diazoalkanes with Terminal Alkynes. J. Am. Chem. Soc. 2016, 138, 14558–14561. [Google Scholar] [CrossRef]
- Zhang, J.; Huo, X.; Xiao, J.; Zhao, L.; Ma, S.; Zhang, W. Enantio- and Diastereodivergent Construction of 1,3-Nonadjacent Stereocenters Bearing Axial and Central Chirality through Synergistic Pd/Cu Catalysis. J. Am. Chem. Soc. 2021, 143, 12622–12632. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Wang, B.; Mu, H.; Zhang, H.; Song, Y.; Qu, J. Development of Tartaric Acid Derived Chiral Guanidines and Their Application to Catalytic Enantioselective α-Hydroxylation of β-Dicarbonyl Compounds. Org. Lett. 2013, 15, 3106–3109. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Bao, X.; Zhang, H.; Song, Y.; Qu, J.; Wang, B. Novel Tartrate-Based Guanidines for Enantioselective Fluorination of 1,3-Dicarbonyl and α-Cyano Carbonyl Compounds. Aust. J. Chem. 2014, 67, 1115–1118. [Google Scholar] [CrossRef]
- Witzig, R.M.; Fäseke, V.C.; Häussinger, D.; Sparr, C. Atroposelective synthesis of tetra-ortho-substituted biaryls by catalyst-controlled non-canonical polyketide cyclizations. Nat. Catal. 2019, 2, 925–930. [Google Scholar] [CrossRef]
- Song, S.; Ma, S. Highly Selective Nucleophilic 4-Aryl-2,3-allenylation of Malonates†. Chin. J. Chem. 2020, 38, 1233–1238. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, X.; Ma, S. Stretchable chiral pockets for palladium-catalyzed highly chemo- and enantioselective allenylation. Nat. Commun. 2021, 12, 2416. [Google Scholar] [CrossRef]
- Fernández, M. Studies on the Chemistry of 2-[3-(2-Nitrophenyl)-2-oxopropyl] benzaldehydes: Novel Syntheses of 5H-Benzo [b] carbazole-6, 11-diones and Indolo [1, 2-b] isoquinoline-6, 11-diones. Synthesis 2009, 2009, 3051–3060. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
 | ||||||||
| Entry a | Ligand | Base | Sol. | T [°C] | t [h] | Yield [%] b | dr c | ee [%] d |
| 1 | L1 | Cs2CO3 | DCM | 25 | 0.5 | 84 | 10:1 | −71/−39 |
| 2 | L2 | Cs2CO3 | DCM | 25 | 12 | trace | - | - |
| 3 | L3 | Cs2CO3 | DCM | 25 | 3 | 89 | 3:1 | −67/−69 |
| 4 | L4 | Cs2CO3 | DCM | 25 | 12 | 55 | 15:1 | −73/−17 |
| 5 | L5 | Cs2CO3 | DCM | 25 | 48 | 21 | 4:1 | −23/−33 |
| 6 | L6 | Cs2CO3 | DCM | 25 | 24 | 20 | 11:1 | −7/−5 |
| 7 | L7 | Cs2CO3 | DCM | 25 | 7.5 | 59 | 5:1 | −69/−69 |
| 8 | L8 | Cs2CO3 | DCM | 25 | 24 | 29 | 5:1 | −89/−51 |
| 9 | L9 | Cs2CO3 | DCM | 25 | 24 | 90 | 4:1 | 91/81 |
| 10 e | L9 | Cs2CO3 | DCM | 25 | 6 | 97 | 4:1 | 93/77 |
| 11 e | L9 | Cs2CO3 | CHCl3 | 25 | 9 | 92 | 5:1 | 92/61 |
| 12 e | L9 | Cs2CO3 | DCE | 25 | 12 | 92 | 4:1 | 91/75 |
| 13 e | L9 | Cs2CO3 | MeCN | 25 | 12 | 96 | 5:1 | 92/81 |
| 14 e | L9 | Cs2CO3 | Tol. | 25 | 9 | 98 | 5:1 | 92/73 |
| 15 e | L9 | Cs2CO3 | THF | 25 | 6 | 97 | 6:1 | 92/65 |
| 16 e | L9 | Cs2CO3 | Dioxane | 25 | 6 | 98 | 4:1 | 93/61 |
| 17 e | L9 | Et3N | THF | 25 | 10 | 70 | 6:1 | 94/71 |
| 18 e | L9 | C4H9OK | THF | 25 | 4 | 90 | 7:1 | 92/71 |
| 19 e | L9 | C2H5ONa | THF | 25 | 6 | 98 | 3:1 | 90/73 |
| 20 e | L9 | NaHCO3 | THF | 25 | 6 | 98 | 7:1 | 93/67 |
| 21 e | L9 | Na2CO3 | THF | 25 | 10 | 61 | 5:1 | 92/51 |
| 22 e | L9 | K2CO3 | THF | 25 | 6 | 98 | 2:1 | 93/79 |
| 23 e | L9 | NaHCO3 | THF | 0 | 24 | 98 | 6:1 | 95/79 |
| 24 e | L9 | NaHCO3 | THF | −10 | 42 | 98 | 7:1 | 95/70 |
| 25 e,f | L9 | NaHCO3 | THF | −10 | 12 | 98 | 7:1 | 96/71 |
| 26 e,g | L9 | NaHCO3 | THF | −10 | 48 | 98 | 8:1 | 96/77 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, A.; Wei, X.; Huang, Y.; Qu, J.; Wang, B. Palladium-Catalyzed Stereoselective Construction of 1,3-Stereocenters Displaying Axial and Central Chirality via Asymmetric Alkylations. Molecules 2023, 28, 2927. https://doi.org/10.3390/molecules28072927
Xue A, Wei X, Huang Y, Qu J, Wang B. Palladium-Catalyzed Stereoselective Construction of 1,3-Stereocenters Displaying Axial and Central Chirality via Asymmetric Alkylations. Molecules. 2023; 28(7):2927. https://doi.org/10.3390/molecules28072927
Chicago/Turabian StyleXue, Aiqi, Xingfu Wei, Yue Huang, Jingping Qu, and Baomin Wang. 2023. "Palladium-Catalyzed Stereoselective Construction of 1,3-Stereocenters Displaying Axial and Central Chirality via Asymmetric Alkylations" Molecules 28, no. 7: 2927. https://doi.org/10.3390/molecules28072927