
Ensemble Learning, Deep Learning-Based and Molecular Descriptor-Based Quantitative Structure–Activity Relationships

{kind=link}
{kind=link}
Abstract
:1. Introduction
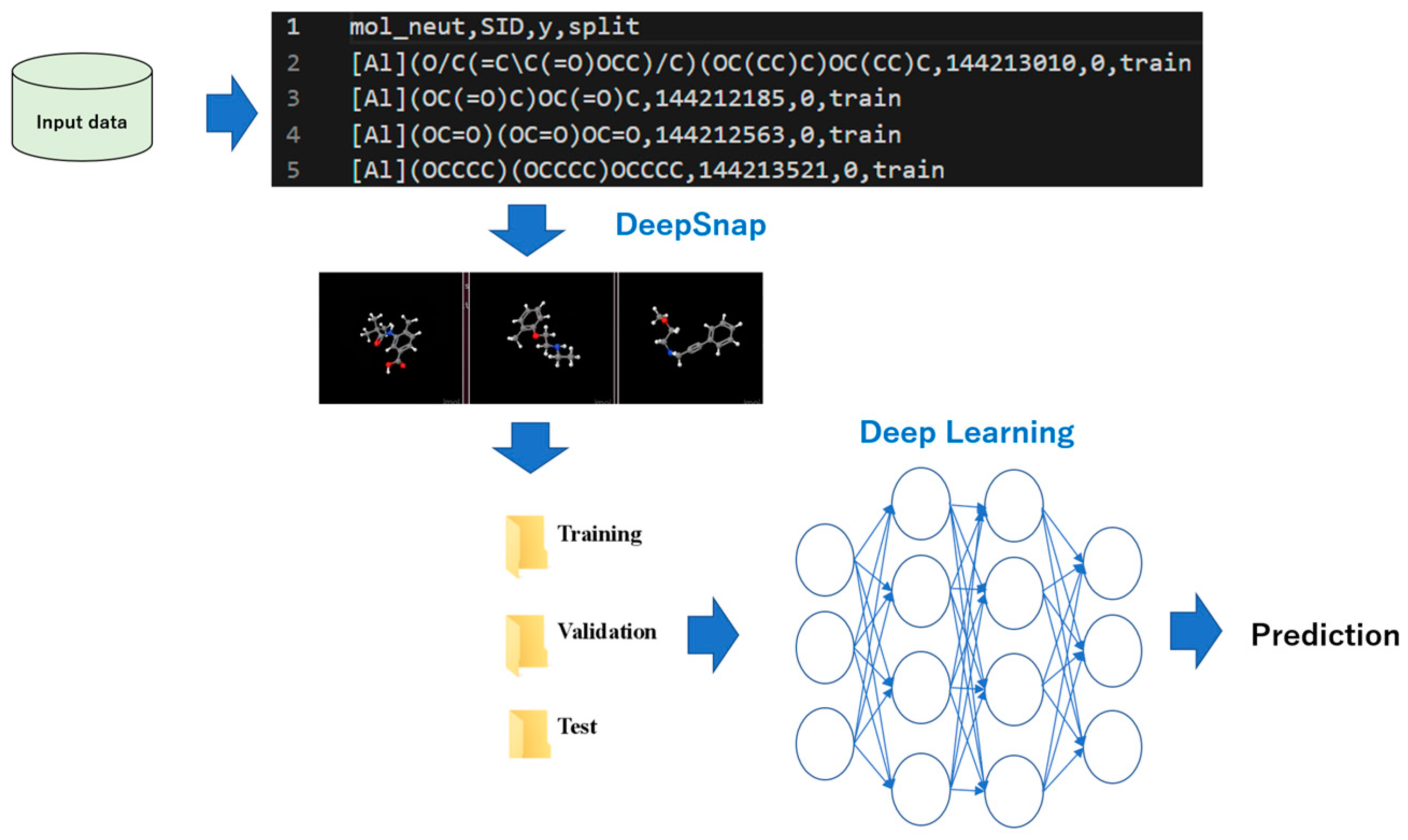
2. Classification of Images by a Neural Network
3. Evaluation of Predictive Models and Predictive Performance
4. Ensemble Learning
- Repeat the following steps B times.
- Create a new dataset by m-time split sampling from the training data.
- Build a weak learner h based on the divided dataset.
- Construct the final learning result using B times weak learners h.Classification: H(x) = arg max |{i|hi = y}|
5. DeepSNAP: DL and EL
6. Application of DL in New Drug Development and Medicine
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Karlin, E.A.; Lin, C.C.; Meftah, M.; Slover, J.D.; Schwarzkopf, R. The Impact of Machine Learning on Total Joint Arthroplasty Patient Outcomes: A Systemic Review. J. Arthroplast. 2022. [Google Scholar] [CrossRef]
- Sourlos, N.; Wang, J.; Nagaraj, Y.; van Ooijen, P.; Vliegenthart, R. Possible Bias in Supervised Deep Learning Algorithms for CT Lung Nodule Detection and Classification. Cancers 2022, 14, 3867. [Google Scholar] [CrossRef]
- Jeng, F.C.; Jeng, Y.S. Implementation of Machine Learning on Human Frequency-Following Responses: A Tutorial. Semin. Hear. 2022, 43, 251–274. [Google Scholar] [CrossRef]
- Ingrosso, A.; Goldt, S. Data-driven emergence of convolutional structure in neural networks. Proc. Natl. Acad. Sci. USA 2022, 119, e2201854119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, F.; Song, X.; Zhou, H.; Yang, Y.; Zhang, H.; Liu, J. A rotation meanout network with invariance for dermoscopy image classification and retrieval. Comput. Biol. Med. 2022, 151, 106272. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y. Deep Neural Networks for QSAR. Methods Mol. Biol. 2022, 2390, 233–260. [Google Scholar] [CrossRef] [PubMed]
- Kaveh, M.; Mesgari, M.S. Application of Meta-Heuristic Algorithms for Training Neural Networks and Deep Learning Architectures: A Comprehensive Review. Neural Process Lett. 2022, in press. [CrossRef]
- Yates, L.; Aandahl, Z.; Richards, S.A.; Brook, B.W. Cross validation for model selection: A primer with examples from ecology. arXiv 2022, arXiv:2203.04552v1. Available online: https://arxiv.org/abs/2203.04552 (accessed on 9 March 2022).
- Cao, Y.; Chen, Z.; Belkin, M.; Gu, Q. Benign Overfitting in Two-layer Convolutional Neural Networks. arXiv 2022, arXiv:2202.06526v3. Available online: https://arxiv.org/abs/2202.06526 (accessed on 14 February 2022).
- Hou, C.K.J.; Behdinan, K. Dimensionality Reduction in Surrogate Modeling: A Review of Combined Methods. Data Sci. Eng. 2022, 4, 402–427. [Google Scholar] [CrossRef]
- Kukačka, J.; Golkov, V.; Cremers, D. Regularization for Deep Learning: A Taxonomy. arXiv 2017, arXiv:1710.10686v1. Available online: https://arxiv.org/abs/1710.10686 (accessed on 29 October 2017).
- Raschka, S. Model Evaluation, Model Selection, and Algorithm Selection in Machine Learning. arXiv 2018, arXiv:1811.12808v33. Available online: https://arxiv.org/abs/1811.12808 (accessed on 13 November 2018).
- Dehghani, A.; Glatard, T.; Shihab, E. Subject Cross Validation in Human Activity Recognition. arXiv 2019, arXiv:1904.02666v2. Available online: https://arxiv.org/abs/1904.02666 (accessed on 4 April 2019).
- Battey, H.S.; Reid, N. Inference in High-dimensional Linear Regression. arXiv 2021, arXiv:2106.12001v2. Available online: https://arxiv.org/abs/2106.12001 (accessed on 22 June 2021).
- Brannath, W.; Scharpenberg, M. Interpretation of Linear Regression Coefficients under Mean Model Miss-Specification. arXiv 2014, arXiv:1409.8544v4. Available online: https://arxiv.org/abs/1409.8544 (accessed on 30 September 2014).
- Gutknecht, A.J.; Barnett, L. Sampling distribution for single-regression Granger causality estimators. arXiv 2018, arXiv:1911.09625v2. Available online: https://arxiv.org/abs/1911.09625 (accessed on 21 November 2019). [CrossRef]
- Schultheiss, C.; Bühlmann, P. Ancestor regression in linear structural equation models. arXiv 2022, arXiv:2205.08925v2. Available online: https://arxiv.org/abs/2205.08925 (accessed on 18 May 2022). [CrossRef]
- Yevkin, G.; Yevkin, O. On regression analysis with Padé approximants. arXiv 2022, arXiv:2208.09945v1. Available online: https://arxiv.org/abs/2208.09945 (accessed on 21 August 2022).
- Choi, J.-E.; Shin, D.W. Quantile correlation coefficient: A new tail dependence measure. arXiv 2018, arXiv:1803.06200v1. Available online: https://arxiv.org/abs/1803.06200 (accessed on 16 May 2018). [CrossRef]
- O’Neill, B. Multiple Linear Regression and Correlation: A Geometric Analysis. arXiv 2021, arXiv:2109.08519v1. Available online: https://arxiv.org/abs/2109.08519 (accessed on 13 September 2021).
- Gupta, I.; Mittal, H.; Rikhari, D.; Singh, A.K. MLRM: A Multiple Linear Regression based Model for Average Temperature Prediction of A Day. arXiv 2022, arXiv:2203.05835v1. Available online: https://arxiv.org/abs/2203.05835 (accessed on 11 March 2022).
- Rocks, J.W.; Mehta, P. Bias-variance decomposition of overparameterized regression with random linear features. Phys. Rev. E 2022, 106, 025304. [Google Scholar] [CrossRef]
- Gao, J. Bias-variance decomposition of absolute errors for diagnosing regression models of continuous data. Patterns 2021, 2, 100309. [Google Scholar] [CrossRef] [PubMed]
- Voncken, L.; Albers, C.J.; Timmerman, M.E. Bias-Variance Trade-Off in Continuous Test Norming. Assessment 2021, 28, 1932–1948. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, J. Mitigating Bias and Error in Machine Learning to Protect Sports Data. Comput. Intell. Neurosci. 2022, 2022, 4777010. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Dimiccoli, M.; Lim, B.Y. Debiased-CAM to mitigate systematic error with faithful visual explanations of machine learning. arXiv 2022, arXiv:2201.12835v2. Available online: https://arxiv.org/abs/2201.12835 (accessed on 30 January 2022).
- Bashir, D.; Montanez, G.D.; Sehra, S.; Segura, P.P.; Lauw, J. An Information-Theoretic Perspective on Overfitting and Underfitting. arXiv 2020, arXiv:2010.06076v2. Available online: https://arxiv.org/abs/2010.06076 (accessed on 12 October 2020).
- Li, Z.; Liu, L.; Dong, C.; Shang, J. Overfitting or Underfitting? Understand Robustness Drop in Adversarial Training. arXiv 2020, arXiv:2010.08034v1. Available online: https://arxiv.org/abs/2010.08034 (accessed on 15 October 2020).
- Zhu, X.; Hu, J.; Xiao, T.; Huang, S.; Wen, Y.; Shang, D. An interpretable stacking ensemble learning framework based on multi-dimensional data for real-time prediction of drug concentration: The example of olanzapine. Front. Pharmacol. 2022, 13, 975855. [Google Scholar] [CrossRef]
- Suri, J.S.; Bhagawati, M.; Paul, S.; Protogerou, A.D.; Sfikakis, P.P.; Kitas, G.D.; Khanna, N.N.; Ruzsa, Z.; Sharma, A.M.; Saxena, S.; et al. A Powerful Paradigm for Cardiovascular Risk Stratification Using Multiclass, Multi-Label, and Ensemble-Based Machine Learning Paradigms: A Narrative Review. Diagnostics 2022, 12, 722. [Google Scholar] [CrossRef]
- Ghiasi, M.M.; Zendehboudi, S. Application of decision tree-based ensemble learning in the classification of breast cancer. Comput. Biol. Med. 2021, 128, 104089. [Google Scholar] [CrossRef] [PubMed]
- Ghojogh, B.; Crowley, M. The Theory Behind Overfitting, Cross Validation, Regularization, Bagging, and Boosting: Tutorial. arXiv 2019, arXiv:1905.12787v1. Available online: https://arxiv.org/abs/1905.12787 (accessed on 28 May 2019).
- Chang, O.; Yao, Y.; Williams-King, D.; Lipson, H. Ensemble Model Patching: A Parameter-Efficient Variational Bayesian Neural Network. arXiv 2019, arXiv:1905.09453v1. Available online: https://arxiv.org/abs/1905.09453 (accessed on 23 May 2019).
- Kumar, R.; Subbiah, G. Zero-Day Malware Detection and Effective Malware Analysis Using Shapley Ensemble Boosting and Bagging Approach. Sensors 2022, 22, 2798. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.H.; Lane, H.Y. A bagging ensemble machine learning framework to predict overall cognitive function of schizophrenia patients with cognitive domains and tests. Asian J. Psychiatr. 2022, 69, 103008. [Google Scholar] [CrossRef] [PubMed]
- Ngo, G.; Beard, R.; Chandra, R. Evolutionary bagging for ensemble learning. arXiv 2022, arXiv:2208.02400v3. Available online: https://arxiv.org/abs/2208.02400 (accessed on 4 August 2022). [CrossRef]
- Song, H.; Dong, C.; Zhang, X.; Wu, W.; Chen, C.; Ma, B.; Chen, F.; Chen, C.; Lv, X. Rapid identification of papillary thyroid carcinoma and papillary microcarcinoma based on serum Raman spectroscopy combined with machine learning models. Photodiagn. Photodyn. Ther. 2022, 37, 102647. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Cai, Y.; Zhao, K.; Xie, H.; Chen, X. Concepts and applications of chemical fingerprint for hit and lead screening. Drug Discov. Today 2022, 27, 103356. [Google Scholar] [CrossRef]
- Bamisile, O.; Cai, D.; Oluwasanmi, A.; Ejiyi, C.; Ukwuoma, C.C.; Ojo, O.; Mukhtar, M.; Huang, Q. Comprehensive assessment, review, and comparison of AI models for solar irradiance prediction based on different time/estimation intervals. Sci. Rep. 2022, 12, 9644. [Google Scholar] [CrossRef]
- Zhao, X.; Lu, Y.; Li, S.; Guo, F.; Xue, H.; Jiang, L.; Wang, Z.; Zhang, C.; Xie, W.; Zhu, F. Predicting renal function recovery and short-term reversibility among acute kidney injury patients in the ICU: Comparison of machine learning methods and conventional regression. Ren. Fail. 2022, 44, 1326–1337. [Google Scholar] [CrossRef]
- Uesawa, Y. Quantitative structure-activity relationship analysis using deep learning based on a novel molecular image input technique. Bioorg. Med. Chem. Lett. 2018, 28, 3400–3403. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, Y.; Uesawa, Y. A Deep Learning-Based Quantitative Structure-Activity Relationship System Construct Prediction Model of Agonist and Antagonist with High Performance. Int. J. Mol. Sci. 2022, 23, 2141. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Totoki, S.; Handa, K.; Shiota, T.; Kurosaki, K.; Uesawa, Y. Prediction Models for Agonists and Antagonists of Molecular Initiation Events for Toxicity Pathways Using an Improved Deep-Learning-Based Quantitative Structure-Activity Relationship System. Int. J. Mol. Sci. 2021, 22, 10821. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, Y.; Uesawa, Y. A Molecular Image-Based Novel Quantitative Structure-Activity Relationship Approach, Deepsnap-Deep Learning and Machine Learning. Curr. Issues Mol. Biol. 2021, 42, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, Y.; Uesawa, Y. Molecular Image-Based Prediction Models of Nuclear Receptor Agonists and Antagonists Using the DeepSnap-Deep Learning Approach with the Tox21 10K Library. Molecules 2020, 25, 2764. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Hosaka, T.; Ogaito, A.; Yoshinari, K.; Uesawa, Y. Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap-Deep Learning. Molecules 2020, 25, 1317. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaka, Y.; Uesawa, Y. DeepSnap-Deep Learning Approach Predicts Progesterone Receptor Antagonist Activity with High Performance. Front. Bioeng. Biotechnol. 2020, 7, 485. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaka, Y.; Uesawa, Y. Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library. Int. J. Mol. Sci. 2019, 20, 4855. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaka, Y.; Uesawa, Y. Optimization of a Deep-Learning Method Based on the Classification of Images Generated by Parameterized Deep Snap a Novel Molecular-Image-Input Technique for Quantitative Structure-Activity Relationship (QSAR) Analysis. Front. Bioeng. Biotechnol. 2019, 7, 65. [Google Scholar] [CrossRef] [Green Version]
- Mamada, H.; Nomura, Y.; Uesawa, Y. Prediction Model of Clearance by a Novel Quantitative Structure-Activity Relationship Approach, Combination DeepSnap-Deep Learning and Conventional Machine Learning. ACS Omega 2021, 6, 23570–23577. [Google Scholar] [CrossRef]
- Mamada, H.; Nomura, Y.; Uesawa, Y. Novel QSAR Approach for a Regression Model of Clearance That Combines DeepSnap-Deep Learning and Conventional Machine Learning. ACS Omega 2022, 7, 17055–17062. [Google Scholar] [CrossRef] [PubMed]
- Daghighi, A.; Casanola-Martin, G.M.; Timmerman, T.; Milenković, D.; Lučić, B.; Rasulev, B. In Silico Prediction of the Toxicity of Nitroaromatic Compounds: Application of Ensemble Learning QSAR Approach. Toxics 2022, 10, 746. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Tanaka, K.; Kotera, M.; Funatsu, K. Comparison and improvement of the predictability and interpretability with ensemble learning models in QSPR applications. J. Cheminform. 2020, 12, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2019, 35, 309–318. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuzaka, Y.; Uesawa, Y. Ensemble Learning, Deep Learning-Based and Molecular Descriptor-Based Quantitative Structure–Activity Relationships. Molecules 2023, 28, 2410. https://doi.org/10.3390/molecules28052410
Matsuzaka Y, Uesawa Y. Ensemble Learning, Deep Learning-Based and Molecular Descriptor-Based Quantitative Structure–Activity Relationships. Molecules. 2023; 28(5):2410. https://doi.org/10.3390/molecules28052410
Chicago/Turabian StyleMatsuzaka, Yasunari, and Yoshihiro Uesawa. 2023. "Ensemble Learning, Deep Learning-Based and Molecular Descriptor-Based Quantitative Structure–Activity Relationships" Molecules 28, no. 5: 2410. https://doi.org/10.3390/molecules28052410