
Targeting Epigenetic Changes Mediated by Members of the SMYD Family of Lysine Methyltransferases


Abstract
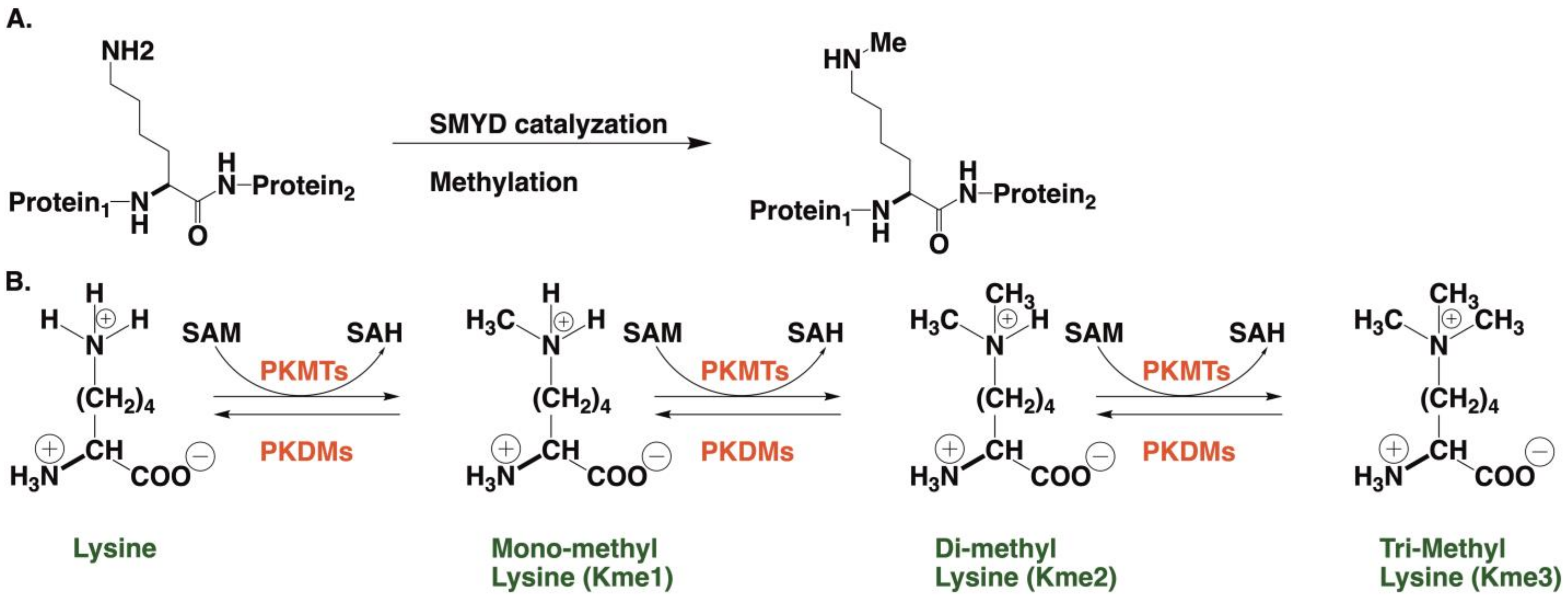
:1. Introduction
2. Approaches to the Development of Pharmacologic Agents Modulating SMYD Function
2.1. Inhibitor Development Based on Structure–Activity Relationships (SAR)
2.2. In Silico Methods for Inhibitor Identification to Structurally Known Proteins
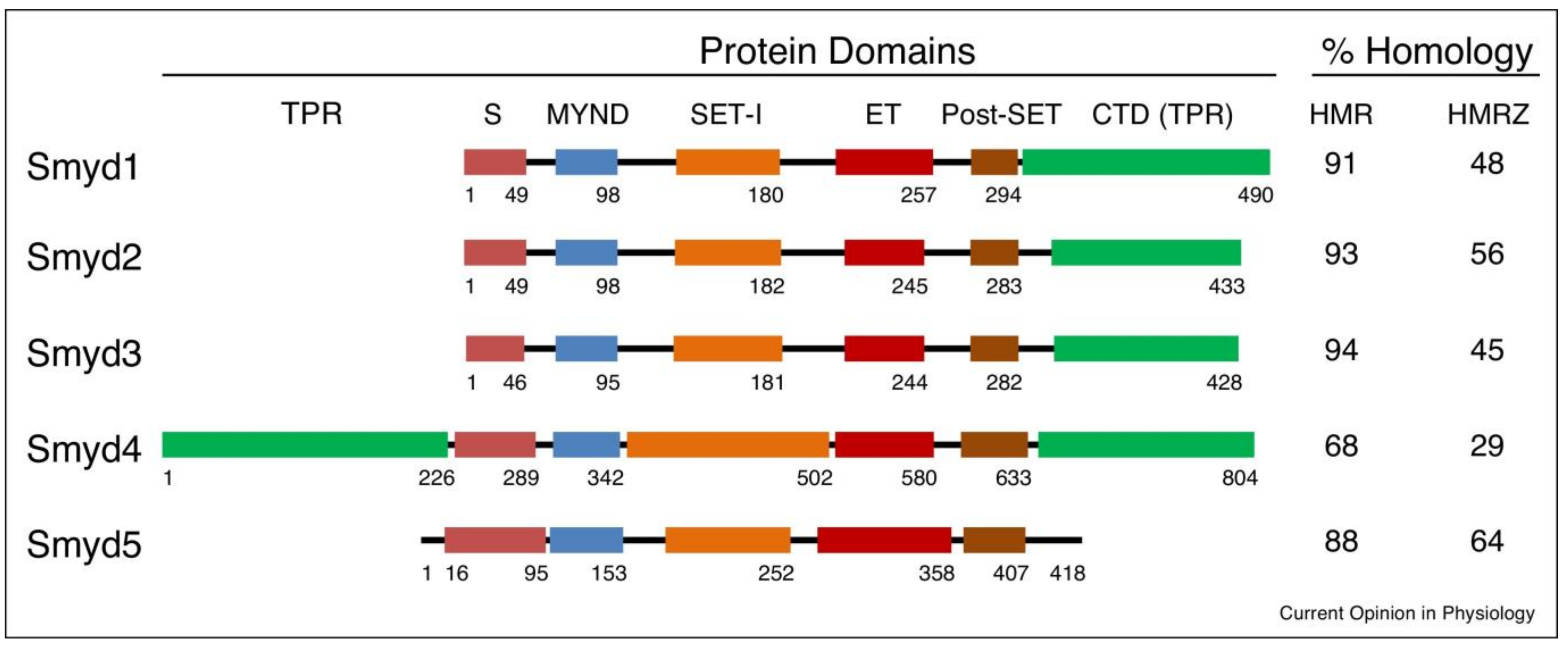
3. SMYD-Proteins, Structure, Function, and Medicinal Potential
3.1. SMYD1
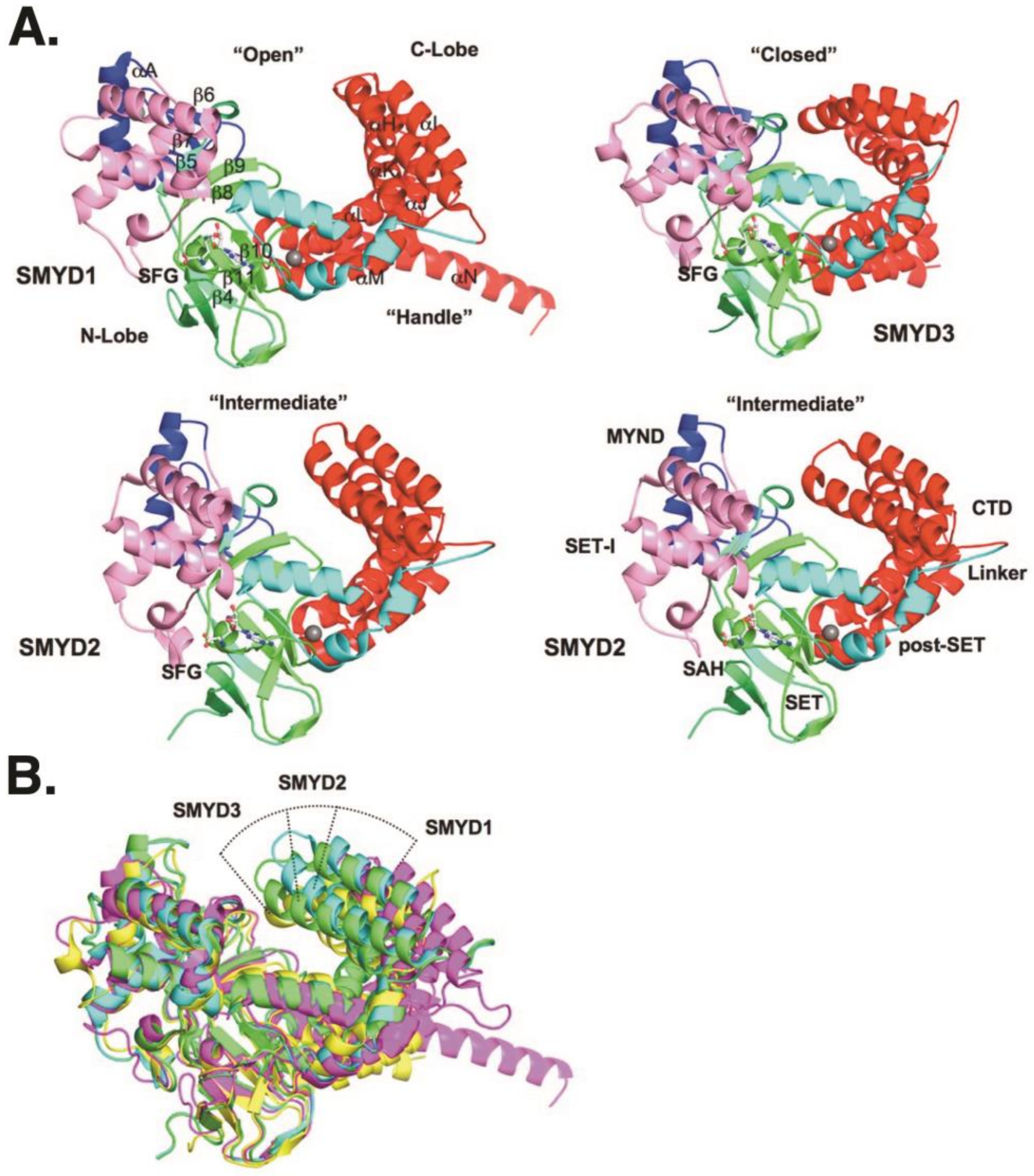
3.1.1. Structure and Function of SMYD1
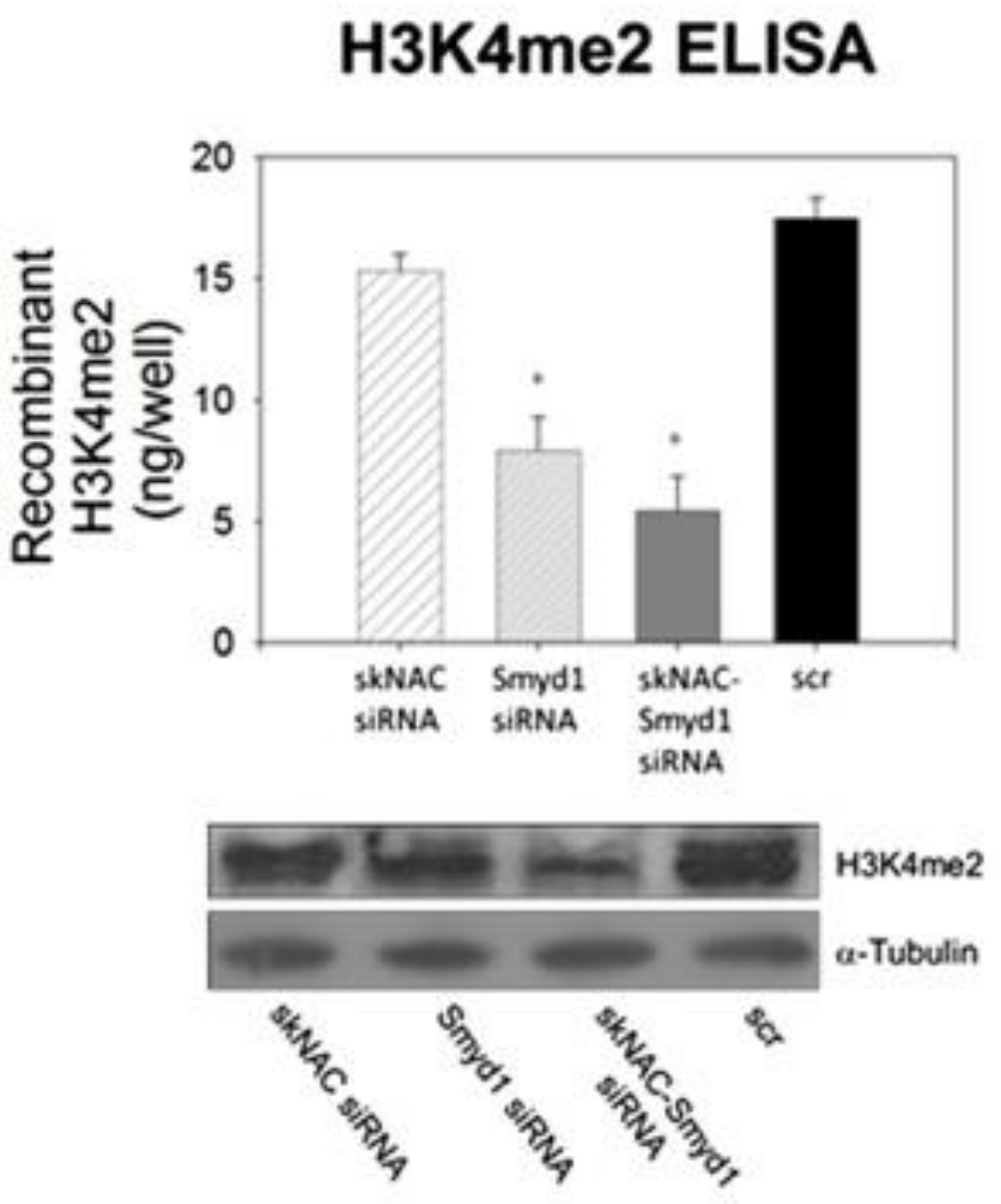
3.1.2. Therapeutic Applications, Potential Drug Targeting, and the Use of SMYD1 as a Prognostic Indicator
3.2. SMYD2
3.2.1. Structure and Function of SMYD2
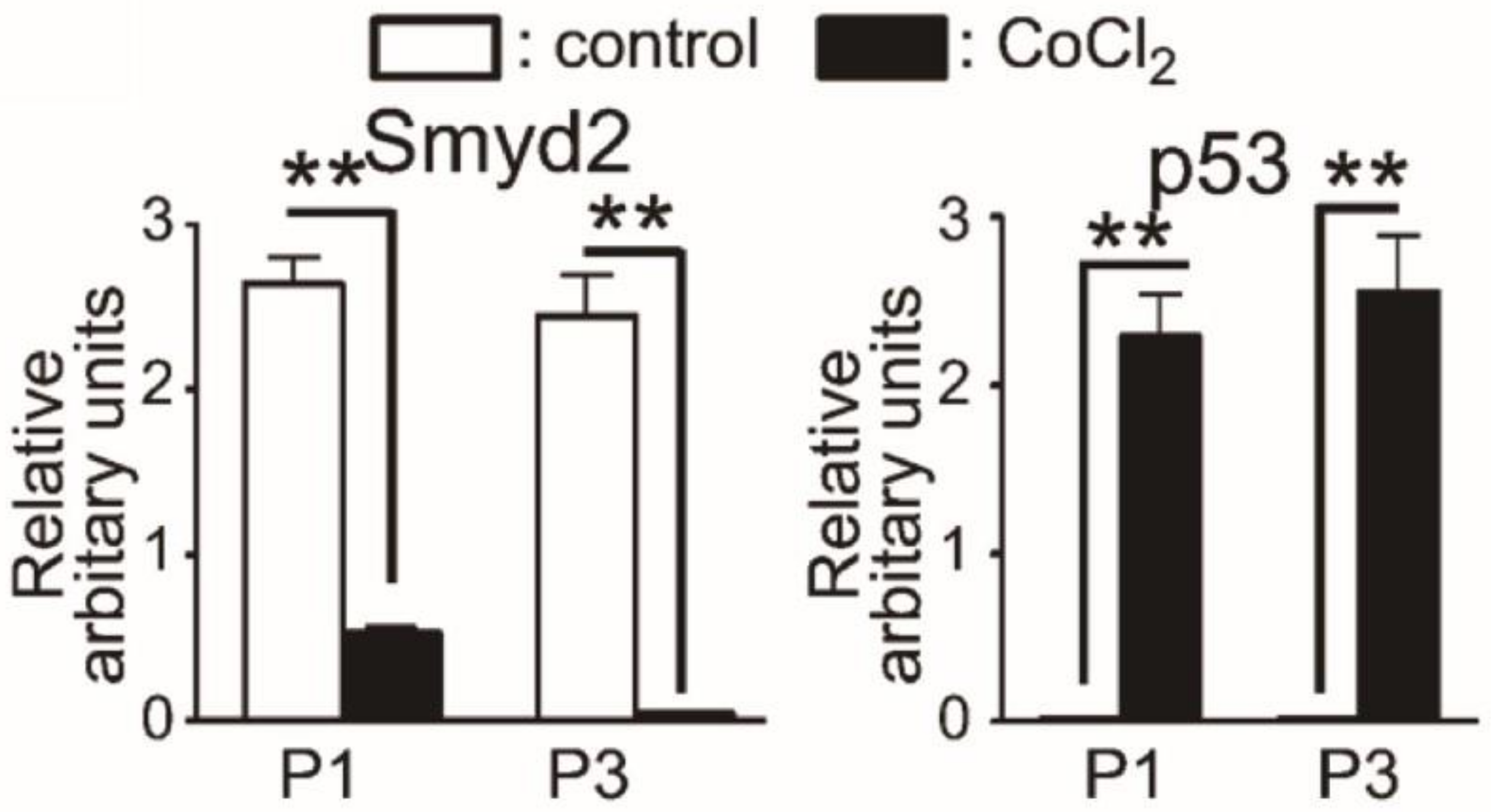
3.2.2. Therapeutic Applications, Potential Drug Targeting, and Prognostics SMYD2
3.3. SMYD3
3.3.1. Structure and Function of SMYD3
3.3.2. Therapeutic Applications, Potential Drug Targeting of SMYD3 and the Prognostic Value of SMYD3
3.4. SMYD4
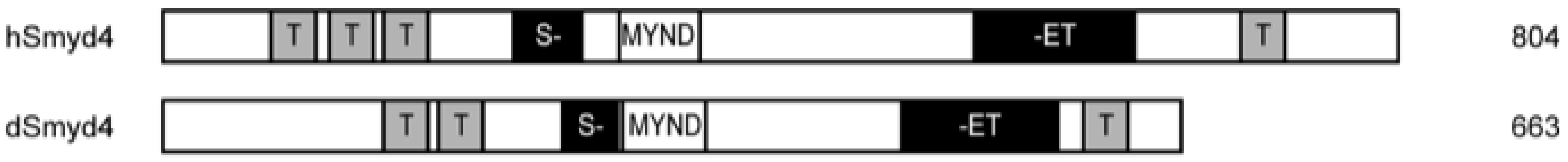
3.4.1. Structure and Function of SMYD4
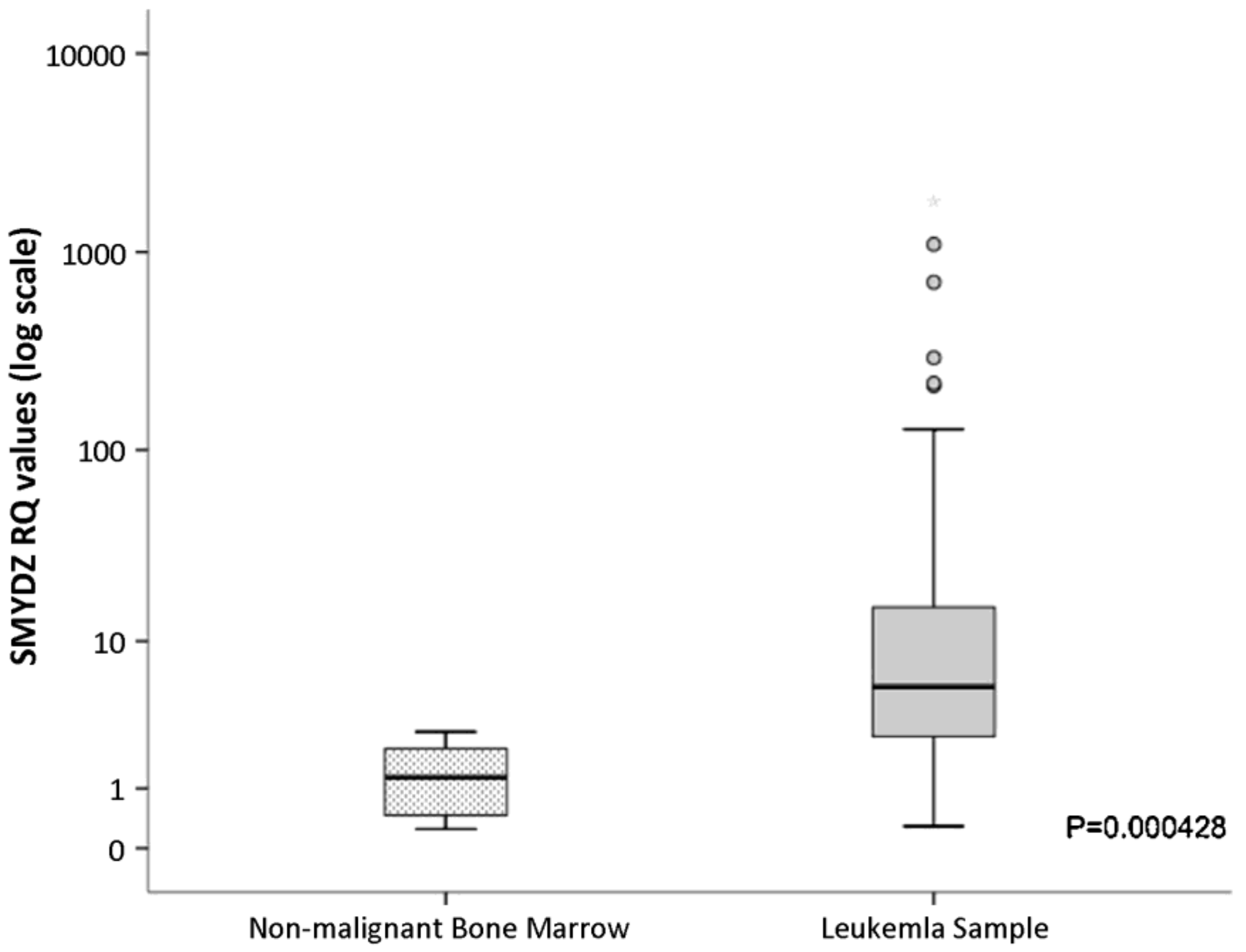
3.4.2. Therapeutic Applications, Potential Drug Targeting and Prognostics of SMYD4
3.5. SMYD5
Structure and Function of SMYD5
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Leinhart, K.; Brown, M. SET/MYND Lysine Methyltransferases Regulate Gene Transcription and Protein Activity. Genes 2011, 2, 210–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, H.J.; Brown, M.A. Characterization of the SMYD Family of Lysine Methyltransferases: Reflections upon Key Findings and Therapeutic Implications. Int. J. Sci. 2015, 4, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Rubio-Tomás, T. The SMYD family proteins in immunology: An update of their obvious and non-obvious relations with the immune system. Heliyon 2021, 7, e07387. [Google Scholar] [CrossRef]
- Aljazi, M.B.; Gao, Y.; Wu, Y.; He, J. SMYD5 is a histone H3-specific methyltransferase mediating mono-methylation of histone H3 lysine 36 and 37. Biochem. Biophys Res. Commun. 2022, 599, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fang, Y.; Tang, Y.; Han, S.; Jia, J.; Wan, X.; Chen, J.; Yuan, Y.; Zhao, B.; Fang, D. SMYD5 catalyzes histone H3 lysine 36 trimethylation at promoters. Nat. Commun. 2022, 13, 3190. [Google Scholar] [CrossRef]
- Tracy, C.; Warren, J.S.; Szulik, M.; Wang, L.; Garcia, J.; Makaju, A.; Russell, K.; Miller, M.; Franklin, S. The Smyd Family of Methyltransferases: Role in Cardiac and Skeletal Muscle Physiology and Pathology. Curr. Opin. Physiol. 2018, 1, 140–152. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Pan, Y.; Ma, X.; Liu, L.; Wang, W.; You, W. SMYD3 overexpression indicates poor prognosis and promotes cell proliferation, migration and invasion in non-small cell lung cancer. Int. J. Oncol. 2020, 57, 756–766. [Google Scholar] [CrossRef]
- Lin, S.Q.; Li, X. Epigenetic Therapies and Potential Drugs for Treating Human Cancer. Curr. Drug Targets 2020, 21, 1068–1083. [Google Scholar] [CrossRef]
- Yi, X.; Jiang, X.J.; Fang, Z.M. Histone methyltransferase SMYD2: Ubiquitous regulator of disease. Clin. Epigenetics 2019, 11, 112. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Lambert, J.-P.; Al-Madhoun, A.S.; Elisma, F.; Skerjanc, I.S.; Figeys, D. The Tale of Two Domains: Proteomics and Genomics Analysis of SMYD2, A New Histone Methyltransferase. Mol. Cell Proteo. 2008, 7, 560–572. [Google Scholar] [CrossRef] [Green Version]
- Bonasio, R.; Zhang, G.; Ye, C.; Mutti, N.S.; Fang, X.; Qin, N.; Donahue, G.; Yang, P.; Li, Q.; Li, C.; et al. Genomic comparison of the ants Camponotus floridanus and Harpegnathos saltator. Science 2010, 329, 1068–1071. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Jin, M.; Jeong, K.W. Histone H3K4 Methyltransferases as Targets for Drug-Resistant Cancers. Biology 2021, 10, 581. [Google Scholar] [CrossRef]
- Kudithipudi, S.; Jeltsch, A. Role of somatic cancer mutations in human protein lysine methyltransferases. Biochim. Biophy Acta 2014, 1846, 366–379. [Google Scholar] [CrossRef]
- Bottino, C.; Peserico, A.; Simone, C.; Caretti, G. SMYD3: An Oncogenic Driver Targeting Epigenetic Regulation and Signaling Pathways. Cancers 2020, 12, 142. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.K. Histone methyl transferases: A class of epigenetic opportunities to counter uncontrolled cell proliferation. Eur. J. Med. Chem. 2019, 166, 351–368. [Google Scholar] [CrossRef]
- Warren, J.S.; Tracy, C.M.; Miller, M.R.; Makaju, A.; Szulik, M.W.; Oka, S.I.; Yuzyuk, T.N.; Cox, J.E.; Kumar, A.; Lozier, B.K.; et al. Histone methyltransferase Smyd1 regulates mitochondrial energetics in the heart. Proc. Natl. Acad. Sci. USA 2018, 115, E7871–E7880. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Schwartz, R.J.; Liu, J.; Sun, F.; Li, Q.; Ma, Y. Smyd1 Orchestrates Early Heart Development Through Positive and Negative Gene Regulation. Front. Cell Dev. Biol. 2021, 9, 654682. [Google Scholar] [CrossRef]
- Nagandla, H.; Lopez, S.; Yu, W.; Rasmussen, T.L.; Tucker, H.O.; Schwartz, R.J.; Stewart, M.D. Defective myogenesis in the absence of the muscle-specific lysine methyltransferase SMYD1. Dev. Biol. 2016, 410, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, L.H.T.; de Andrade, R.V.; Soares Felipe, M.S.; Motoyama, A.B.; Silva, F.P. SMYD2 is highly expressed in pediatric acute lymphoblastic leukemi and constitutes a bad prognotic factor. Leuk. Res. 2014, 38, 496–502. [Google Scholar] [CrossRef]
- Du, S.J.; Tan, X.; Zhang, J. SMYD proteins: Key regulators in skeletal and cardiac muscle development and function. Anat. Rec. 2014, 297, 1650–1662. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kouzarides, T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002, 12, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Sims, R.J., 3rd; Gottlieb, P.D.; Tucker, P.W. Identification and characterization of Smyd2: A split SET/MYND domain-containing histone H3 lysine 36-specific methyltransferase that interacts with the Sin3 histone deacetylase complex. Mol. Cancer 2006, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, D.; Turniansky, R.; Moresco, J.J.; Ikram, S.; Ramaprasad, G.; Akinwole, A.; Wolf, J.; Yates, J.R., 3rd; Green, E.M. Function of the MYND Domain and C-Terminal Region in Regulating the Subcellular Localization and Catalytic Activity of the SMYD Family Lysine Methyltransferase Set5. Mol. Cell Biol. 2020, 40, e00341-19. [Google Scholar] [CrossRef]
- Rister, J.; Desplan, C. Deciphering the genome’s regulatory code: The many languages of DNA. Bioessays 2010, 32, 381–384. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, M.; Sugimoto, K.; Fukushima, T.; Shinkai, Y. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J. Biol. Chem. 2001, 276, 25309–25317. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, X.; He, K.; Ma, Y.; Su, N.; He, H.; Stolc, V.; Tongprasit, W.; Jin, W.; Jiang, J.; et al. High-resolution mapping of epigenetic modifications of the rice genome uncovers interplay between DNA methylation, histone methylation, and gene expression. Plant Cell 2008, 20, 259–276. [Google Scholar] [CrossRef] [Green Version]
- Rueda-Robles, A.; Audano, M.; Alvarez-Mercado, A.I.; Rubio-Tomas, T. Functions of SMYD proteins in biological processes: What do we know? An updated review. Arch. Biochem. Biophys. 2021, 712, 109040. [Google Scholar] [CrossRef]
- Spellmon, N.; Holcomb, J.; Trescott, L.; Sirinupong, N.; Yang, Z. Structure and function of SET and MYND domain-containing proteins. Int. J. Mol. Sci. 2015, 16, 1406–1428. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.A.; Foreman, K.; Harriss, J.; Das, C.; Zhu, L.; Edwards, M.; Shaaban, S.; Tucker, H. C-terminal domain of SMYD3 serves as a unique HSP90-regulated motif in oncogenesis. Oncotarget 2015, 6, 4005–4019. [Google Scholar] [CrossRef] [Green Version]
- Foreman, K.W.; Brown, M.; Park, F.; Emtage, S.; Harriss, J.; Das, C.; Zhu, L.; Crew, A.; Arnold, L.; Shaaban, S.; et al. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS ONE 2011, 6, e22290. [Google Scholar] [CrossRef]
- Jarrell, D.K.; Hassell, K.N.; Alshiraihi, I.; Crans, D.C.; Brown, M.A. Structural Analysis of SMYD3 Lysine Methyltransferase for the Development of Competitive and Specific Enzyme Inhibitors. Diseases 2022, 10, 4. [Google Scholar] [CrossRef]
- Borlak, J.; Thum, T. Hallmarks of ion channel gene expression in end-stage heart failure. FASEB J. 2003, 17, 1592–1608. [Google Scholar] [CrossRef] [Green Version]
- Li, L.X.; Zhou, J.X.; Calvet, J.P.; Godwin, A.K.; Jensen, R.A.; Li, X. Lysine methyltransferase SMYD2 promotes triple negative breast cancer progression. Cell Death Dis. 2018, 9, 326. [Google Scholar] [CrossRef] [Green Version]
- Hamamoto, R.; Furukawa, Y.; Morita, M.; Iimura, Y.; Silva, F.P.; Li, M.; Yagyu, R.; Nakamura, Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat. Cell Biol. 2004, 6, 731–740. [Google Scholar] [CrossRef]
- Luo, X.G.; Zhang, C.L.; Zhao, W.W.; Liu, Z.P.; Liu, L.; Mu, A.; Guo, S.; Wang, N.; Zhou, H.; Zhang, T.C. Histone methyltransferase SMYD3 promotes MRTF-A-mediated transactivation of MYL9 and migration of MCF-7 breast cancer cells. Cancer Lett. 2014, 344, 129–137. [Google Scholar] [CrossRef]
- Wang, S.Z.; Luo, X.G.; Shen, J.; Zou, J.N.; Lu, Y.H.; Xi, T. Knockdown of SMYD3 by RNA interference inhibits cervical carcinoma cell growth and invasion in vitro. BMB Rep. 2008, 41, 294–299. [Google Scholar] [CrossRef]
- Vieira, F.Q.; Costa-Pinheiro, P.; Ramalho-Carvalho, J.; Pereira, A.; Menezes, F.D.; Antunes, L.; Carneiro, I.; Oliveira, J.; Henrique, R.; Jerónimo, C. Deregulated expression of selected histone methylases and demethylases in prostate carcinoma. Endocr. Relat. Cancer 2014, 21, 51–61. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Xu, J.; Zhang, J.; Xie, D.; Ye, H.; Xiao, Z.; Cai, M.; Xu, K.; Zeng, Y.; Li, H.; et al. High expression of trimethylated histone H3 lysine 4 is associated with poor prognosis in hepatocellular carcinoma. Hum. Pathol. 2012, 43, 1425–1435. [Google Scholar] [CrossRef]
- Sponziello, M.; Durante, C.; Boichard, A.; Dima, M.; Puppin, C.; Verrienti, A.; Tamburrano, G.; Di Rocco, G.; Redler, A.; Lacroix, L.; et al. Epigenetic-related gene expression profile in medullary thyroid cancer revealed the overexpression of the histone methyltransferases EZH2 and SMYD3 in aggressive tumours. Mol. Cell Endocrinol. 2014, 392, 8–13. [Google Scholar] [CrossRef]
- Pan, K.; Hu, B.; Wang, L.; Yuan, J.; Xu, W. STUB1-SMYD2 Axis Regulates Drug Resistance in Glioma cells. J. Mol. Neurosci. 2022, 72, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Peserico, A.; Germani, A.; Sanese, P.; Barbosa, A.J.; Di Virgilio, V.; Fittipaldi, R.; Fabini, E.; Bertucci, C.; Varchi, G.; Moyer, M.P.; et al. A SMYD3 Small-Molecule Inhibitor Impairing Cancer Cell Growth. J. Cell Physiol. 2015, 230, 2447–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshiraihi, I.M.; Jarrell, D.K.; Arhouma, Z.; Hassell, K.N.; Montgomery, J.; Padilla, A.; Ibrahim, H.M.; Crans, D.C.; Kato, T.A.; Brown, M.A. In Silico/In Vitro Hit-to-Lead Methodology Yields SMYD3 Inhibitor That Eliminates Unrestrained Proliferation of Breast Carcinoma Cells. Int. J. Mol. Sci. 2020, 21, 9549. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liew, S.S.; Lin, G.R.; Poulsen, A.; Ang, M.J.Y.; Chia, B.C.S.; Chew, S.Y.; Kwek, Z.P.; Wee, J.L.K.; Ong, E.H.; et al. Discovery of Irreversible Inhibitors Targeting Histone Methyltransferase, SMYD3. ACS Med. Chem. Lett. 2019, 10, 978–984. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Larsen, N.A.; Howard, T.; Pollard, H.; Green, I.; Grande, C.; Cheung, T.; Garcia-Arenas, R.; Cowen, S.; Wu, J.; et al. Structural basis of substrate methylation and inhibition of SMYD2. Structure 2011, 19, 1262–1273. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, L.H.; Boriack-Sjodin, P.A.; Smith, S.; Thomenius, M.; Rioux, N.; Munchhof, M.; Mills, J.E.; Klaus, C.; Totman, J.; Riera, T.V.; et al. Novel Oxindole Sulfonamides and Sulfamides: EPZ031686, the First Orally Bioavailable Small Molecule SMYD3 Inhibitor. ACS Med. Chem. Lett. 2016, 7, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Raj, K.; Mufti, G.J. Azacytidine (Vidaza(R)) in the treatment of myelodysplastic syndromes. Ther. Clin. Risk Manag. 2006, 2, 377–388. [Google Scholar] [CrossRef] [Green Version]
- Nieto, M.; Demolis, P.; Béhanzin, E.; Moreau, A.; Hudson, I.; Flores, B.; Stemplewski, H.; Salmonson, T.; Gisselbrecht, C.; Bowen, D.; et al. The European Medicines Agency Review of Decitabine (Dacogen) for the Treatment of Adult Patients With Acute Myeloid Leukemia: Summary of the Scientific Assessment of the Committee for Medicinal Products for Human Use. Oncologist 2016, 21, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Bubna, A.K. Vorinostat-An Overview. Indian J. Derm. 2015, 60, 419. [Google Scholar] [CrossRef]
- VanderMolen, K.M.; McCulloch, W.; Pearce, C.J.; Oberlies, N.H. Romidepsin (Istodax, NSC 630176, FR901228, FK228, depsipeptide): A natural product recently approved for cutaneous T-cell lymphoma. J. Antibiot. 2011, 64, 525–531. [Google Scholar] [CrossRef]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Wang, T.; Geng, C.; Zhang, Y.; Zhang, J.; Ning, Z.; Jiang, Z. Exploratory clinical study of chidamide, an oral subtype-selective histone deacetylase inhibitor, in combination with exemestane in hormone receptor-positive advanced breast cancer. Chin. J. Cancer Res. 2018, 30, 605–612. [Google Scholar] [CrossRef]
- Sweis, R.F.; Pliushchev, M.; Brown, P.J.; Guo, J.; Li, F.; Maag, D.; Petros, A.M.; Soni, N.B.; Tse, C.; Vedadi, M.; et al. Discovery and Development of Potent and Selective Inhibitors of Histone Methyltransferase G9a. ACS Medl. Chem. Lett. 2014, 5, 205–209. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.; Allali-Hassani, A.; Antonysamy, S.; Chang, S.; Chen, L.H.; Curtis, C.; Emtage, S.; Fan, L.; Gheyi, T.; Li, F.; et al. LLY-507, a Cell-active, Potent, and Selective Inhibitor of Protein-lysine Methyltransferase SMYD2. J. Biol. Chem. 2015, 290, 13641–13653. [Google Scholar] [CrossRef] [Green Version]
- Aureliano, M.; Gumerova, N.I.; Sciortino, G.; Garribba, E.; Rompel, A.; Crans, D.C. Polyoxovanadates with emerging biomedical activities. Coord. Chem. Rev. 2021, 447, 214143. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Hu, L.; Zhu, Y.T.; Qi, C.; Zhu, Y.J. Identification of Smyd4 as a potential tumor suppressor gene involved in breast cancer development. Cancer Res. 2009, 69, 4067–4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulton, M.D.; Brown, T.; Zheng, Y.G. Mechanisms and inhibitors of histone arginine methylation. Chem. Record 2018, 18, 1792–1807. [Google Scholar] [CrossRef]
- Al-Shar’i, N.A.; Alnabulsi, S.M. Explaining the autoinhibition of the SMYD enzyme family: A theoretical study. J. Mol. Graph Model 2016, 68, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Sirinupong, N.; Brunzelle, J.; Ye, J.; Pirzada, A.; Nico, L.; Yang, Z. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture. J. Biol. Chem. 2010, 285, 40635–40644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Sirinupong, N.; Brunzelle, J.; Yang, Z. Crystal structures of histone and p53 methyltransferase SmyD2 reveal a conformational flexibility of the autoinhibitory C-terminal domain. PLoS ONE 2011, 6, e21640. [Google Scholar] [CrossRef]
- Xu, S.; Zhong, C.; Zhang, T.; Ding, J. Structure of human lysine methyltransferase Smyd2 reveals insights into the substrate divergence in Smyd proteins. J. Mol. Cell Biol. 2011, 3, 293–300. [Google Scholar] [CrossRef] [Green Version]
- Dashti, P.; van de Peppel, J.; Thaler, R.; Paradise, C.R.; Stein, G.S.; Montecino, M.A.; van Leeuwen, J.P.T.M.; van der Eerden, B.J.; Dudakovic, A.; van Wijnen, A.J. The lysine methyltransferases SET and MYND domain containing 2 (Smyd2) and Enhancer of Zeste 2 (Ezh2) co-regulate osteoblast proliferation and mineralization. Gene 2023, 851, 146928. [Google Scholar] [CrossRef]
- Sirinupong, N.; Brunzelle, J.; Doko, E.; Yang, Z. Structural insights into the autoinhibition and posttranslational activation of histone methyltransferase SmyD3. J. Mol. Biol. 2011, 406, 149–159. [Google Scholar] [CrossRef]
- Wang, K.Z.; Zhu, X.; Li, Y.L.; Chen, D.X.; Wu, P.; Chu, W.Y. Molecular characterization and expression regulation of Smyd1a and Smyd1b in skeletal muscle of Chinese perch (Siniperca chuatsi). Comp. Biochem. Phys. B- Biochem. Mol. Biol. 2016, 194, 25–31. [Google Scholar] [CrossRef]
- Zhang, Y.; Alshammari, E.; Sobota, J.; Yang, A.; Li, C.; Yang, Z. Unique SMYD5 Structure Revealed by AlphaFold Correlates with Its Functional Divergence. Biomolecules 2022, 12, 783. [Google Scholar] [CrossRef]
- Hanf, A.; Oelze, M.; Manea, A.; Li, H.; Munzel, T.; Daiber, A. The anti-cancer drug doxorubicin induces substantial epigenetic changes in cultured cardiomyocytes. Chem. Biol. Interact. 2019, 313, 108834. [Google Scholar] [CrossRef]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, eaal3321. [Google Scholar] [CrossRef]
- Jarrell, D.K.; Hassell, K.N.; Crans, D.C.; Lanning, S.; Brown, M.A. Characterizing the Role of SMYD2 in Mammalian Embryogenesis-Future Directions. Vet. Sci. 2020, 7, 63. [Google Scholar] [CrossRef]
- Voelkel, T.; Andresen, C.; Unger, A.; Just, S.; Rottbauer, W.; Linke, W.A. Lysine methyltransferase Smyd2 regulates Hsp90-mediated protection of the sarcomeric titin springs and cardiac function. Biochim. Biophys. Acta 2013, 1833, 812–822. [Google Scholar] [CrossRef]
- Brown, M.A.; Edwards, M.A.; Alshiraihi, I.; Geng, H.; Dekker, J.D.; Tucker, H.O. The lysine methyltransferase SMYD2 is required for normal lymphocyte development and survival of hematopoietic leukemias. Genes Immun. 2020, 21, 119–130. [Google Scholar] [CrossRef]
- Diehl, F.; Brown, M.A.; van Amerongen, M.J.; Novoyatleva, T.; Wietelmann, A.; Harriss, J.; Ferrazzi, F.; Bottger, T.; Harvey, R.P.; Tucker, P.W.; et al. Cardiac deletion of Smyd2 is dispensable for mouse heart development. PLoS ONE 2010, 5, e9748. [Google Scholar] [CrossRef] [Green Version]
- Thompson, E.C.; Travers, A.A. A Drosophila Smyd4 homologue is a muscle-specific transcriptional modulator involved in development. PLoS ONE 2008, 3, e3008. [Google Scholar] [CrossRef] [Green Version]
- Sims, R.J.; Reinberg, D. From chromatin to cancer: A new histone lysine methyltransferase enters the mix. Nat. Cell Biol. 2004, 6, 685–687. [Google Scholar] [CrossRef]
- Komatsu, S.; Ichikawa, D.; Hirajima, S.; Nagata, H.; Nishimura, Y.; Kawaguchi, T.; Miyamae, M.; Okajima, W.; Ohashi, T.; Konishi, H.; et al. Overexpression of SMYD2 contributes to malignant outcome in gastric cancer. Br. J. Cancer 2015, 112, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Doughan, M. SMYD4 Subcellular Localization and Nogo-B Receptor Structure and Function. Master’s Theses, Wayne State University, Detroit, MI, USA, 2018; p. 732. [Google Scholar]
- Xiao, D.; Wang, H.; Hao, L.; Guo, X.; Ma, X.; Qian, Y.; Chen, H.; Ma, J.; Zhang, J.; Sheng, W.; et al. The roles of SMYD4 in epigenetic regulation of cardiac development in zebrafish. PLoS Genet. 2018, 14, e1007578. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.J.; Xu, P.F.; Zhou, T.; Hu, M.; Fu, C.T.; Zhang, Y.; Jin, Y.; Chen, Y.; Chen, S.J.; Huang, Q.H.; et al. Genome-wide survey and developmental expression mapping of zebrafish SET domain-containing genes. PLoS ONE 2008, 3, e1499. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Aizaki, Y.; Sato, K.; Oda, H.; Kurokawa, R.; Mimura, T. Altered gene expression profiles of histone lysine methyltransferases and demethylases in rheumatoid arthritis synovial fibroblasts. Clin. Exp. Rheumatol. 2018, 36, 314–316. [Google Scholar] [PubMed]
- Chi, G.; Pei, J.; Li, X.; Li, X.; Pang, H.; Cui, J.; Wu, D.; Qu, G.; He, Y. SMYD5 acts as a potential biomarker for hepatocellular carcinoma. Exp. Cell Res. 2022, 414, 113076. [Google Scholar] [CrossRef] [PubMed]
- Kidder, B.L.; Hu, G.; Cui, K.; Zhao, K. SMYD5 regulates H4K20me3-marked heterochromatin to safeguard ES cell self-renewal and prevent spurious differentiation. Epigen Chroma 2017, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidder, B.L.; He, R.; Wangsa, D.; Padilla-Nash, H.M.; Bernardo, M.M.; Sheng, S.; Ried, T.; Zhao, K. SMYD5 Controls Heterochromatin and Chromosome Integrity during Embryonic Stem Cell Differentiation. Cancer Res. 2017, 77, 6729–6745. [Google Scholar] [CrossRef] [Green Version]
- Stender, J.D.; Pascual, G.; Liu, W.; Kaikkonen, M.U.; Do, K.; Spann, N.J.; Boutros, M.; Perrimon, N.; Rosenfeld, M.G.; Glass, C.K. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol. Cell 2012, 48, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Fujii, T.; Tsunesumi, S.; Sagara, H.; Munakata, M.; Hisaki, Y.; Sekiya, T.; Furukawa, Y.; Sakamoto, K.; Watanabe, S. Smyd5 plays pivotal roles in both primitive and definitive hematopoiesis during zebrafish embryogenesis. Sci. Rep. 2016, 6, 29157. [Google Scholar] [CrossRef] [Green Version]
- Jiao, S.; Xu, R.; Du, S. Smyd1 is essential for myosin expression and sarcomere organization in craniofacial, extraocular, and cardiac muscles. J. Genet. Genom. 2021, 48, 208–218. [Google Scholar] [CrossRef]
- Just, S.; Meder, B.; Berger, I.M.; Etard, C.; Trano, N.; Patzel, E.; Hassel, D.; Marquart, S.; Dahme, T.; Vogel, B.; et al. The myosin-interacting protein SMYD1 is essential for sarcomere organization. J. Cell Sci. 2011, 124, 3127–3136. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.L.; Ma, Y.; Park, C.Y.; Harriss, J.; Pierce, S.A.; Dekker, J.D.; Valenzuela, N.; Srivastava, D.; Schwartz, R.J.; Stewart, M.D.; et al. Smyd1 facilitates heart development by antagonizing oxidative and ER stress responses. PLoS ONE 2015, 10, e0121765. [Google Scholar] [CrossRef] [Green Version]
- Franklin, S.; Kimball, T.; Rasmussen, T.L.; Rosa-Garrido, M.; Chen, H.; Tran, T.; Miller, M.R.; Gray, R.; Jiang, S.; Ren, S.; et al. The chromatin-binding protein Smyd1 restricts adult mammalian heart growth. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1234–H1247. [Google Scholar] [CrossRef] [Green Version]
- Berkholz, J.; Orgeur, M.; Stricker, S.; Munz, B. skNAC and Smyd1 in transcriptional control. Exp. Cell Res. 2015, 336, 182–191. [Google Scholar] [CrossRef]
- Chen, M.; Li, J.; Wang, J.; Le, Y.; Liu, C. SMYD1 alleviates septic myocardial injury by inhibiting endoplasmic reticulum stress. Biosci. Biotechnol. Biochem. 2021, 85, 2383–2391. [Google Scholar] [CrossRef]
- Chow, M.Z.; Sadrian, S.N.; Keung, W.; Geng, L.; Ren, L.; Kong, C.W.; Wong, A.O.; Hulot, J.S.; Chen, C.S.; Costa, K.D.; et al. Modulation of chromatin remodeling proteins SMYD1 and SMARCD1 promotes contractile function of human pluripotent stem cell-derived ventricular cardiomyocyte in 3D-engineered cardiac tissues. Sci. Rep. 2019, 9, 7502. [Google Scholar] [CrossRef] [Green Version]
- Mayfield, R.D.; Zhu, L.; Smith, T.A.; Tiwari, G.R.; Tucker, H.O. The SMYD1 and skNAC transcription factors contribute to neurodegeneratie diseases. Brain Behav. Immun.—Health 2020, 9, 100129. [Google Scholar] [CrossRef]
- Yan, L.; Ding, B.; Liu, H.; Zhang, Y.; Zeng, J.; Hu, J.; Yao, W.; Yu, G.; An, R.; Chen, Z.; et al. Inhibition of SMYD2 suppresses tumor progression by down-regulating microRNA-125b and attenuates multi-drug resistance in renal cell carcinoma. Theranostics 2019, 9, 8377–8391. [Google Scholar] [CrossRef]
- Boehm, D.; Jeng, M.; Camus, G.; Gramatica, A.; Schwarzer, R.; Johnson, J.R.; Hull, P.A.; Montano, M.; Sakane, N.; Pagans, S.; et al. SMYD2-Mediated Histone Methylation Contributes to HIV-1 Latency. Cell Host. Microbe 2017, 21, 569–579.e6. [Google Scholar] [CrossRef] [Green Version]
- Abu-Farha, M.; Lanouette, S.; Elisma, F.; Tremblay, V.; Butson, J.; Figeys, D.; Couture, J.F. Proteomic analyses of the SMYD family interactomes identify HSP90 as a novel target for SMYD2. J. Mol. Cell Biol. 2011, 3, 301–308. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Zhang, D.; Lian, M. Identification of an epigenetic prognostic signature for patients with lower-grade gliomas. CNS Neurosci. Ther. 2021, 27, 470–483. [Google Scholar] [CrossRef]
- Donlin, L.T.; Andresen, C.; Just, S.; Rudensky, E.; Pappas, C.T.; Kruger, M.; Jacobs, E.Y.; Unger, A.; Zieseniss, A.; Dobenecker, M.W.; et al. Smyd2 controls cytoplasmic lysine methylation of Hsp90 and myofilament organization. Genes Dev. 2012, 26, 114–119. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.A.; Brown, M.A.; Alshiraihi, I.; Jarrell, D.K.; Tucker, H.O. The Lysine Methyltransferase SMYD2 Is Required for Definite Hematopoietic Stem Cell Production in the Mouse Embryo. Vet. Sci. 2020, 7, 100. [Google Scholar] [CrossRef]
- Sajjad, A.; Novoyatleva, T.; Vergarajauregui, S.; Troidl, C.; Schermuly, R.T.; Tucker, H.O.; Engel, F.B. Lysine methyltransferase Smyd2 suppresses p53-dependent cardiomyocyte apoptosis. Biochim. Biophys. Acta 2014, 1843, 2556–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.C.; Shan, S.K.; Guo, B.; Li, C.C.; Li, F.X.; Zheng, M.H.; Xu, Q.S.; Wang, Y.; Lei, L.M.; Tang, K.X.; et al. Histone Lysine Methylation Modification and Its Role in Vascular Calcification. Front. Endocrinol. 2022, 13, 863708. [Google Scholar] [CrossRef]
- Chen, J.; Gu, Y.L.; Zhang, H.; Zhang, N.Y.; Song, N.; Hu, J.C.; Cai, J.R.; Shi, Y.Q.; Ding, X.Q.; Zhang, X.Y. Amelioration of uremic toxin indoxyl slfate-indced osteroblastic calcification by SET domain containing lysne methytransferase 7/9 protein. Nephron 2019, 141, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Song, M.K.; Jung, S.; Hong, S.; Kwon, J.O.; Kim, M.K.; Kim, H.H. Effects of the Lysine Methyltransferase Inhibitor AZ505 on Bone Metabolism. J. Bone Metab. 2021, 28, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Trescott, L.; Holcomb, J.; Zhang, X.; Brunzelle, J.; Sirinupong, N.; Shi, X.; Yang, Z. Structural insights into estrogen receptor a methylation by histone methyltransferase SMYD2, a cellular event impliated in estrogen signaling regulation. J. Mol. Biol. 2014, 426, 3413–3425. [Google Scholar] [CrossRef]
- Shang, L.; Wei, M. Inhibition of SMYD2 Sensitized Cisplatin to Resistant Cells in NSCLC Through Activating p53 Pathway. Front. Oncol. 2019, 9, 306. [Google Scholar] [CrossRef]
- Ren, H.; Wang, Z.; Chen, Y.; Liu, Y.; Zhang, S.; Zhang, T.; Li, Y. SMYD2-OE promotes oxaliplatin resistance in colon cancer through MDR1/P-glycoprotein via MEK/ERK/AP1 pathway. Onco. Targets Ther. 2019, 12, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Siddique, A.B.; Ebrahim, H.Y.; Tajmim, A.; King, J.A.; Abdelwahed, K.S.; Abd Elmageed, Z.Y.; El Sayed, K.A. Oleocanthal Attenuates Metastatic Castration-Resistant Prostate Cancer Progression and Recurrence by Targeting SMYD2. Cancers 2022, 14, 3542. [Google Scholar] [CrossRef]
- Van der Hage, J.A.; van de Velde, C.J.; Julien, J.P.; Tubiana-Hulin, M.; Vandervelden, C.; Duchateau, L. Preoperative chemotherapy in primary operable breast cancer: Results from the European Organization for Research and Treatment of Cancer trial 10902. J. Clin. Oncol. 2001, 19, 4224–4237. [Google Scholar] [CrossRef]
- Sobral, R.A.; Honda, S.T.; Katayama, M.L.; Brentani, H.; Brentani, M.M.; Patrao, D.F.; Folgueira, M.A. Tumor slices as a model to evaluate doxorubicin in vitro treatment and expression of trios of genes PRSS11, MTSS1, CLPTM1 and PRSS11, MTSS1, SMYD2 in canine mammary gland cancer. Acta Vet. Scand. 2008, 50, 27. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Reynoird, N.; Khatri, P.; Jansen, P.W.; Wilkinson, A.W.; Liu, S.; Barbash, O.; Van Aller, G.S.; Huddleston, M.; Dhanak, D.; et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature 2014, 510, 283–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colon-Bolea, P.; Crespo, P. Lysine methylation in cancer: SMYD3-MAP3K2 teaches us new lessons in the Ras-ERK pathway. Bioessays 2014, 36, 1162–1169. [Google Scholar] [CrossRef] [PubMed]
- Sawicka-Gutaj, N.; Shawkat, S.; Andrusiewicz, M.; Ziolkowska, P.; Czarnywojtek, A.; Gut, P.; Ruchala, M. EZH2 and SMYD3 expression in papillary thyroid cancer. Oncol. Lett. 2021, 21, 342. [Google Scholar] [CrossRef] [PubMed]
- Gradl, S.; Steuber, H.; Weiske, J.; Szewczyk, M.M.; Schmees, N.; Siegel, S.; Stoeckigt, D.; Christ, C.D.; Li, F.; Organ, S.; et al. Discovery of the SMYD3 Inhibitor BAY-6035 Using Thermal Shift Assay (TSA)-Based High-Throughput Screening. SLAS Discov. 2021, 26, 947–960. [Google Scholar] [CrossRef]
- Su, D.-S.; Qu, J.; Schulz, M.; Blackledge, C.W.; Yu, H.; Zeng, J.; Burgess, J.; Reif, A.; Stern, M.; Nagarajan, R.; et al. Discovery of Isoxazole Amides as Potent and Selective SMYD3 Inhibitors. ACS Med. Chem. Lett. 2020, 11, 133–140. [Google Scholar] [CrossRef]
- Eggert, E.; Hillig, R.C.; Koehr, S.; Stöckigt, D.; Weiske, J.; Barak, N.; Mowat, J.; Brumby, T.; Christ, C.D.; Ter Laak, A.; et al. Discovery and Characterization of a Highly Potent and Selective Aminopyrazoline-Based in Vivo Probe (BAY-598) for the Protein Lysine Methyltransferase SMYD2. J. Med. Chem. 2016, 59, 4578–4600. [Google Scholar] [CrossRef] [Green Version]
- Parenti, M.D.; Naldi, M.; Manoni, E.; Fabini, E.; Cederfelt, D.; Talibov, V.O.; Gressani, V.; Guven, U.; Grossi, V.; Fasano, C.; et al. Discovery of the 4-aminopiperidine-based compound EM127 for the site-specific covalent inhibition of SMYD3. Eur. J. Med. Chem. 2022, 243, 114683. [Google Scholar] [CrossRef]
- Sarris, M.E.; Moulos, P.; Haroniti, A.; Giakountis, A.; Talianidis, I. Smyd3 Is a Transcriptional Potentiator of Multiple Cancer-Promoting Genes and Required for Liver and Colon Cancer Development. Cancer Cell 2016, 29, 354–366. [Google Scholar] [CrossRef] [Green Version]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Saberianpour, S.; Abkhooie, L. MiR-1307: A comprehensive review of its role in various cancer. Gene Rep. 2021, 25, 101392. [Google Scholar] [CrossRef]
- Han, S.; Zou, H.; Lee, J.W.; Han, J.; Kim, H.C.; Cheol, J.J.; Kim, L.S.; Kim, H. miR-1307-3p Stimulates Breast Cancer Development and Progression by Targeting SMYD4. J. Cancer 2019, 10, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Boriack-Sjodin, P.A.; Swinger, K.K. Protein Methyltransferases: A Distinct, Diverse, and Dynamic Family of Enzymes. Biochemistry 2016, 55, 1557–1569. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme and Date of Discovery | AA Length | X-ray Structure Solved | Regulating Cellular Pathways | Histone Substrate | Inhibitors | Linked to Diseases | Special Features |
|---|---|---|---|---|---|---|---|
| SMYD1 1995 | 472 [2,6] | Yes SET divided by MYND [29,45,64,65,66,67] | Reduced methylation of cardio endoplasmic reticulum stress metabolic sensor, TRB3, [16,17,18] cardiomyocyte differentiation. Muscle-specific for cardiac function. | H3k4, H3K9, H3K27, and H4K20 [1,26] H9c2 rat myoblasts [71] | Breast cancer [72] Cardiac diseases [17] | Multiple isoforms in cardiac and skeletal muscle tissue [16,18] Early heart development [17] Prostate cancer [19] | |
| SMYD2 | 433 [6] | Yes SET divided by MYND [29,45,64,65,66,67] | Negative regulator of macrophage activation, MI polarization. Lowered inerleukin-6 (IL-6), tumor necrosis factor alpha (TNFα) and other proinflammatory cytokine differentiation of the mesendoderm [73,74]. SMYD2 as an oncogene [75]. | H3K4 methyltransferase related to H3K4me2,3 H3K36 [76,77]. | AZ50552 [45] A-89361 [54] LLY-50762 [55] | Liver, kidneys, thymus, hypothalamus, vomeronasal organ (VNO), and ovaries [78] Liver hematomas Leukemias such as CML, ALL, B-ALL, MLLr-B-ALL, AML, and T-ALL [75] gastric cancers [79] | Selective for lysine methylation on histone and non-histone proteins such as retinoblastoma protein (RB) and tumor suppressors (p53) [73,74] |
| SMYD3 | SMYD3 | Yes [29,31,68,69] | Heart and skeletal development, as well as multiple dysfunctional health pathways. [68] | H3K4 Hepatocellular, colon, and breast carcinoma regulator [31] | BCI-121, EPZ031686, EPZ030456, EPZ028862, GSK-49, BAY-6035. EP2031686 [31] | Hepatocellular malignancies [31] | Establish a method for trimethylation of genetic sequences [43] |
| SMYD4 | 854 [6] | No [29]. | Tumor suppression [6,61,80] | Unknown [6] | Unknown [6] | Zebrafish [81,82] using Drosophila SMYD4 (dSMYD4) [77,81] | Congenital heart defects, [81] rheumatoid arthritis [3] autoimmune disease [83] |
| SMYD5 | 4 [6] | No [70,84] | Repression of cytokine transcription of genes [3,4,85]. Self-renewal of embryonic stem cells (ESC) [86]. | H4K20me3 [86] H3K36 and H3K37 [4] H3L36me3 [5] H3K9 [87] | Zebrafish model [88] | Hepatocellular carcinomas [84] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padilla, A.; Manganaro, J.F.; Huesgen, L.; Roess, D.A.; Brown, M.A.; Crans, D.C. Targeting Epigenetic Changes Mediated by Members of the SMYD Family of Lysine Methyltransferases. Molecules 2023, 28, 2000. https://doi.org/10.3390/molecules28042000
Padilla A, Manganaro JF, Huesgen L, Roess DA, Brown MA, Crans DC. Targeting Epigenetic Changes Mediated by Members of the SMYD Family of Lysine Methyltransferases. Molecules. 2023; 28(4):2000. https://doi.org/10.3390/molecules28042000
Chicago/Turabian StylePadilla, Alyssa, John F. Manganaro, Lydia Huesgen, Deborah A. Roess, Mark A. Brown, and Debbie C. Crans. 2023. "Targeting Epigenetic Changes Mediated by Members of the SMYD Family of Lysine Methyltransferases" Molecules 28, no. 4: 2000. https://doi.org/10.3390/molecules28042000