
Computational Exploration of Dirhodium Complex-Catalyzed Selective Intermolecular Amination of Tertiary vs. Benzylic C−H Bonds


Abstract
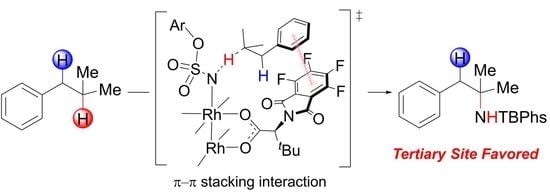
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. The Dirhodium Complex Rh2(S-tfpttl)4
2.2. The Dirhodium–Nitrene Complex
2.3. Singlet Pathway
2.4. Triplet Pathway
2.5. Origins of Site-Selectivity
3. Computational Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gridnev, I.D.; Imamoto, T. On the Mechanism of Stereoselection in Rh-Catalyzed Asymmetric Hydrogenation: A General Approach for Predicting the Sense of Enantioselectivity. Acc. Chem. Res. 2004, 37, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Direct Functionalization of Nitrogen Heterocycles via Rh-Catalyzed C−H Bond Activation. Acc. Chem. Res. 2008, 41, 1013–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collet, F.; Dodd, R.H.; Dauban, P. Catalytic C–H Amination: Recent Progress and Future Directions. Chem. Commun. 2009, 34, 5061–5074. [Google Scholar] [CrossRef] [PubMed]
- Zalatan, D.N.; Du Bois, J. Metal-Catalyzed Oxidations of C−H to C−N Bonds. Top. Curr. Chem. 2009, 292, 347–378. [Google Scholar]
- Park, Y.; Kim, Y.; Chang, S. Transition Metal-Catalyzed C–H Amination: Scope, Mechanism, and Applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Hayashi, H.; Uchida, T. Nitrene Transfer Reactions for Asymmetric C–H Amination: Recent Development. Eur. J. Org. Chem. 2020, 8, 909–916. [Google Scholar] [CrossRef] [Green Version]
- Espino, C.G.; Fiori, K.W.; Kim, M.; Du Bois, J. Expanding the Scope of C–H Amination through Catalyst Design. J. Am. Chem. Soc. 2004, 126, 15378–15379. [Google Scholar] [CrossRef]
- Espino, C.G.; Wehn, P.M.; Chow, J.; Du Bois, J. Synthesis of 1,3-Difunctionalized Amine Derivatives through Selective C−H Bond Oxidation. J. Am. Chem. Soc. 2001, 123, 6935–6936. [Google Scholar] [CrossRef]
- Guthikonda, K.; Du Bois, J. A Unique and Highly Efficient Method for Catalytic Olefin Aziridination. J. Am. Chem. Soc. 2002, 124, 13672–13673. [Google Scholar] [CrossRef]
- Zalatan, D.N.; Du Bois, J. A Chiral Rhodium Carboxamidate Catalyst for Enantioselective C−H Amination. J. Am. Chem. Soc. 2008, 130, 9220–9221. [Google Scholar] [CrossRef] [Green Version]
- Zalatan, D.N.; Du Bois, J. Understanding the Differential Performance of Rh2(esp)2 as a Catalyst for C−H Amination. J. Am. Chem. Soc. 2009, 131, 7558–7559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinman, A.; Du Bois, J. A Stereoselective Synthesis of (−)-Tetrodotoxin. J. Am. Chem. Soc. 2003, 125, 11510–11511. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.J.; Du Bois, J. A Synthesis of (+)-Saxitoxin. J. Am. Chem. Soc. 2006, 128, 3926–3927. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Robert-Peillard, F.; Fruit, C.; Müller, P.; Dodd, R.H.; Dauban, P. Efficient Diastereoselective Intermolecular Rhodium-Catalyzed C−H Amination. Angew. Chem. Int. Ed. 2006, 45, 4641–4644. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Collet, F.; Robert-Peillard, F.; Müller, P.; Dodd, R.H.; Dauban, P. Toward a Synthetically Useful Stereoselective C-H Amination of Hydrocarbons. J. Am. Chem. Soc. 2008, 130, 343–350. [Google Scholar] [CrossRef]
- Singh, R.; Nagesh, K.; Parameshwar, M. Rhodium(II)-Catalyzed Undirected and Selective C(sp2)−H Amination en Route to Benzoxazolones. ACS Catal. 2016, 6, 6520–6524. [Google Scholar] [CrossRef]
- Munnuri, S.; Adebesin, A.M.; Paudyal, M.P.; Yousufuddin, M.; Dalipe, A.; Falck, J.R. Catalyst-Controlled Diastereoselective Synthesis of Cyclic Amines via C−H Functionalization. J. Am. Chem. Soc. 2017, 139, 18288–18294. [Google Scholar] [CrossRef]
- Li, Q.; Liu, W.; Dang, Y. Origins of Ligand-Controlled Diastereoselectivity in Dirhodium-Catalysed Direct Amination of Aliphatic C(sp3)–H Bonds. Catal. Sci. Technol. 2021, 11, 6960–6964. [Google Scholar] [CrossRef]
- Fanourakis, A.; Williams, B.D.; Paterson, K.J.; Phipps, R.J. Enantioselective Intermolecular C−H Amination Directed by a Chiral Cation. J. Am. Chem. Soc. 2021, 143, 10070–10076. [Google Scholar] [CrossRef]
- Suárez, J.R.; Chiara, J.L. Rhodium-Catalyzed intermolecular C–H Amination of Simple Hydrocarbons using the Shelf-Stable Nonafluorobutanesulfonyl Azide. Chem. Commun. 2013, 49, 9194–9196. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Bergsten, T.M.; Groves, J.T. Manganese-Catalyzed Late-Stage Aliphatic C−H Azidation. J. Am. Chem. Soc. 2015, 137, 5300–5303. [Google Scholar] [CrossRef] [PubMed]
- Margrey, K.A.; Czaplyski, W.L.; Nicewicz, D.A.; Alexanian, E.J. A General Strategy for Aliphatic C−H Functionalization Enabled by Organic Photoredox Catalysis. J. Am. Chem. Soc. 2018, 140, 4213–4217. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.-E.; Chen, S.-J.; Mandal, M.; Guzei, I.A.; Cramer, C.J.; Stahl, S.S. Site-Selective Copper-Catalyzed Azidation of Benzylic C−H Bonds. J. Am. Chem. Soc. 2020, 142, 11388–11393. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Jiang, C.; Liang, Y.; Liu, D.; Bu, F.; Shi, R.; Chen, H.; Chowdhury, A.; Lei, A. Manganese-Catalyzed Oxidative Azidation of C(sp3)−H Bonds under Electrophotocatalytic Conditions. J. Am. Chem. Soc. 2020, 142, 17693–17702. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.H.; Samanta, R.C.; Vecchio, A.D.; Ackermann, L. Mangana(III/IV) Electro-Catalyzed C(sp3)−H Azidation. Chem. Sci. 2021, 12, 2890–2897. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Jin, S.; Kim, D.; Hong, S.H.; Chang, S. Cobalt-Catalyzed Intermolecular C−H Amination of Unactivated Alkanes. J. Am. Chem. Soc. 2021, 143, 5191–5200. [Google Scholar] [CrossRef] [PubMed]
- Fiori, K.W.; Espino, C.G.; Brodsky, B.H.; Du Bois, J. A Mechanistic Analysis of the Rh-Catalyzed Intramolecular C–H Amination Reaction. Tetrahedron 2009, 65, 3042–3051. [Google Scholar] [CrossRef]
- Jeffreya, J.L.; Sarpong, R. Intramolecular C(sp3)–H amination. Chem. Sci. 2013, 4, 4092–4106. [Google Scholar] [CrossRef]
- Roizen, J.L.; Harvey, M.E.; Du Bois, J. Metal-Catalyzed Nitrogen-Atom Transfer Methods for the Oxidation of Aliphatic C–H Bonds. Acc. Chem. Res. 2012, 45, 911–922. [Google Scholar] [CrossRef] [Green Version]
- Fiori, K.W.; Du Bois, J. Catalytic Intermolecular Amination of C–H Bonds: Method Development and Mechanistic Insights. J. Am. Chem. Soc. 2007, 129, 562–568. [Google Scholar] [CrossRef]
- Roizen, J.L.; Zalatan, D.N.; Du Bois, J. Selective Intermolecular Amination of C–H Bonds at Tertiary Carbon Center. Angew. Chem. Int. Ed. 2013, 52, 11343–11346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, J.R.; Feng, K.; Sookezian, A.; White, M.C. Manganese-Catalysed Benzylic C(sp3)−H Amination for Late-Stage Functionalization. Nat. Chem. 2018, 10, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Bess, E.N.; DeLuca, R.J.; Tindall, D.J.; Oderinde, M.S.; Roizen, J.L.; Du Bois, J.; Sigman, M.S. Analyzing Site Selectivity in Rh2(esp)2-Catalyzed Intermolecular C–H Amination Reactions. J. Am. Chem. Soc. 2014, 136, 5783–5789. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, C.; Weng, Y.; Xu, H. Insight into the Mechanism and Site-Selectivity of Rh2II,II(esp)2-Catalyzed Intermolecular C–H Amination. Catal. Sci. Technol. 2016, 6, 5292–5303. [Google Scholar] [CrossRef]
- Nasrallah, A.; Lazib, Y.; Boquet, V.; Darses, B.; Dauban, P. Catalytic Intermolecular C(sp3)−H Amination with Sulfamates for the Asymmetric Synthesis of Amines. Org. Process Res. Dev. 2020, 24, 724–728. [Google Scholar] [CrossRef]
- Brunard, E.; Boquet, V.; Elslande, E.V.; Saget, T.; Dauban, P. Catalytic Intermolecular C(sp3)–H Amination: Selective Functionalization of Tertiary C–H Bonds vs Activated Benzylic C–H Bonds. J. Am. Chem. Soc. 2021, 143, 6407–6412. [Google Scholar] [CrossRef]
- Nasrallah, A.; Boquet, V.; Hecker, A.; Retailleau, P.; Darses, B.; Dauban, P. Catalytic Enantioselective Intermolecular Benzylic C(sp3)–H Amination. Angew. Chem. Int. Ed. 2019, 58, 8192–8196. [Google Scholar] [CrossRef]
- Lin, X.; Zhao, C.; Che, C.-M.; Ke, Z.; Phillips, D.L. A DFT Study on the Mechanism of Rh2II,II-Catalyzed Intramolecular Amidation of Carbamates. Chem. Asian J. 2007, 2, 1101–1108. [Google Scholar] [CrossRef]
- Yuan, S.-W.; Han, H.; Li, Y.-L.; Wu, X.; Bao, X.; Gu, Z.-Y.; Xia, J.-B. Intermolecular C−H Amidation of (Hetero)arenes to Produce Amides through Rhodium-Catalyzed Carbonylation of Nitrene Intermediates. Angew. Chem. Int. Ed. 2019, 58, 8887–8892. [Google Scholar] [CrossRef]
- Xu, H.; Zhang, X.; Ke, Z.; Zhao, C. A Theoretical Study of Dirhodium-Catalyzed Intramolecular Aliphatic C–H bond Amination of Aryl Azides. RSC Adv. 2016, 6, 29045–29053. [Google Scholar] [CrossRef]
- Zhang, X.; Ke, Z.; DeYonker, N.J.; Xu, H.; Li, Z.-F.; Xu, X.; Zhang, X.; Su, C.-Y.; Phillips, D.L.; Zhao, C. Mechanism and Enantioselectivity of Dirhodium-Catalyzed Intramolecular C−H Amination of Sulfamate. J. Org. Chem. 2013, 78, 12460–12468. [Google Scholar] [CrossRef] [PubMed]
- Azek, E.; Spitz, C.; Ernzerhof, M.; Lebela, H. A Mechanistic Study of the Stereochemical Outcomes of Rhodium-Catalysed Styrene Aziridinations. Adv. Synth. Catal. 2020, 362, 384–397. [Google Scholar] [CrossRef]
- Jat, J.L.; Paudyal, M.P.; Gao, H.; Xu, Q.-L.; Yousufuddin, M.; Devarajan, D.; Ess, D.H.; Kürti, L.; Falck, J.R. Direct Stereospecific Synthesis of Unprotected N-H and N-Me Aziridines from Olefins. Science 2014, 343, 61–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, K.; Liu, W.; Niemeyer, Z.L.; Ren, Z.; Bacsa, J.; Musaev, D.G.; Sigman, M.S.; Davies, H.M.L. Site-Selective Carbene-Induced C−H Functionalization Catalyzed by Dirhodium Tetrakis(triarylcyclopropanecarboxylate) Complexes. ACS Catal. 2018, 8, 678–682. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Bruzinski, P.R.; Lake, D.H.; Kong, N.; Fal, M.J. Asymmetric Cyclopropanations by Rhodium(II)N-(Arylsulfonyl)prolinate Catalyzed Decomposition of Vinyldiazomethanes in the Presence of Alkenes. Practical Enantioselective Synthesis of the Four Stereoisomers of 2-Phenylcyclopropan-1-amino Acid. J. Am. Chem. Soc. 1996, 118, 6897–6907. [Google Scholar] [CrossRef]
- Lindsay, V.N.G.; Lin, W.; Charette, A.B. Experimental Evidence for the All-Up Reactive Conformation of Chiral Rhodium(II) Carboxylate Catalysts: Enantioselective Synthesis of cis-Cyclopropane α-Amino Acids. J. Am. Chem. Soc. 2009, 131, 16383–16385. [Google Scholar] [CrossRef]
- De Angelis, A.; Dmitrenko, O.; Yap, G.P.A.; Fox, J.M. Chiral Crown Conformation of Rh2(S-PTTL)4: Enantioselective Cyclopropanation with α-Alkyl-α-diazoesters. J. Am. Chem. Soc. 2009, 131, 7230–7231. [Google Scholar] [CrossRef] [Green Version]
- Boquet, V.; Nasrallah, A.; Dana, A.L.; Brunard, E.; Di Chenna, P.H.; Duran, F.J.; Retailleau, P.; Darses, B.; Sircoglou, M.; Dauban, P. Rhodium(II)-Catalyzed Enantioselective Intermolecular Aziridination of Alkenes. J. Am. Chem. Soc. 2022, 144, 17156–17164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, H.; Liu, X.; Phillips, D.L.; Zhao, C. Mechanistic Insight into the Intramolecular Benzylic C–H Nitrene Insertion Catalyzed by Bimetallic Paddlewheel Complexes: Influence of the Metal Centers. Chem. Eur. J. 2016, 22, 7288–7297. [Google Scholar] [CrossRef]
- Ess, D.H.; Houk, K.N. Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc. 2007, 129, 10646–10647. [Google Scholar] [CrossRef]
- Hong, X.; Liang, Y.; Houk, K.N. Mechanisms and Origins of Switchable Chemoselectivity of Ni–Catalyzed C(aryl)-O and C(acyl)-O Activation of Aryl Esters with Phosphine Ligands. J. Am. Chem. Soc. 2014, 136, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-F.; Liang, Y.; Liu, F.; Houk, K.N. Diels-Alder Reactivities of Benzene, Pyridine, and Di-, Tri-, and Tetrazines: The Roles of Geometrical Distortions and Orbital Interactions. J. Am. Chem. Soc. 2016, 138, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liang, Y.; Houk, K.N. Bioorthogonal Cycloadditions: Computational Analysis with the Distortion/Interaction Model and Predictions of Reactivities. Acc. Chem. Res. 2017, 50, 2297–2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.-F.; Hong, X.; Yu, J.-Q.; Houk, K.N. Experimental−Computational Synergy for Selective Pd(II)-Catalyzed C−H Activation of Aryl and Alkyl Groups. Acc. Chem. Res. 2017, 50, 2853–2860. [Google Scholar] [CrossRef]
- Yu, J.-L.; Zhang, S.-Q.; Hong, X. Mechanisms and Origins of Chemo- and Regioselectivities of Ru(II)-Catalyzed Decarboxylative C−H Alkenylation of Aryl Carboxylic Acids with Alkynes: A Computational Study. J. Am. Chem. Soc. 2017, 139, 7224–7243. [Google Scholar] [CrossRef]
- Gutierrez, O.; Hendrick, C.E.; Kozlowski, M.C. Divergent Reactivity in Pd-Catalyzed [3,3]-Sigmatropic Rearrangement of Allyloxy- and Propargyloxyindoles Revealed by Computation and Experiment. Org. Lett. 2018, 20, 6539–6543. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, X.; Li, G.; Chen, Q.; Yang, Y.-F.; She, Y.-B. Computational Exploration of Chiral Iron Porphyrin-Catalyzed Asymmetric Hydroxylation of Ethylbenzene Where Stereoselectivity Arises from π−π Stacking Interaction. J. Org. Chem. 2019, 84, 13755–13763. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Honda, K.T.; Uchimaru, M. Mikami, Ab initio Calculations of Structures and Interaction Energies of Toluene Dimers Including CCSD(T) Level Electron Correlation Correction. J. Chem. Phys. 2005, 122, 144323. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Maseras, F.; Morokuma, K. IMOMM: A New Integrated Ab initio + Molecular Mechanics Geometry Optimization Scheme of Equilibrium Structures and Transition States. J. Comput. Chem. 1995, 16, 1170–1179. [Google Scholar] [CrossRef]
- Svensson, M.; Humbel, S.; Froese, R.D.J.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A Multilayered Integrated MO + MM Method for Geometry Optimizations and Single Point Energy Predictions. A Test for Diels-Alder Reactions and Pt(P(t-Bu)3)2 + H2 Oxidative Addition. J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar] [CrossRef]
- Maseras, F. The IMOMM Method Opens the Way for the Accurate Calculation of “Real” Transition Metal Complexes. Chem. Commun. 2000, 19, 1821–1827. [Google Scholar] [CrossRef]
- Ananikov, V.P.; Szilagyi, R.; Morokuma, K.; Musaev, D.G. Can Steric Effects Induce the Mechanism Switch in the Rhodium-Catalyzed Imine Boration Reaction? A Density Functional and ONIOM Study. Organometallics 2005, 24, 1938–1946. [Google Scholar] [CrossRef]
- Qin, C.; Boyarskikh, V.; Hansen, J.H.; Hardcastle, K.I.; Musaev, D.G.; Davies, H.M.L. D2-Symmetric Dirhodium Catalyst Derived from a 1,2,2-Triarylcyclopropanecarboxylate Ligand: Design, Synthesis and Application. J. Am. Chem. Soc. 2011, 133, 19198–19204. [Google Scholar] [CrossRef] [PubMed]
- McCullough, E.A.; Aprà, E.; Nichols, J. Comparison of the Becke−Lee−Yang−Parr and Becke−Perdew−Wang Exchange-Correlation Functionals for Geometries of Cyclopentadienyl−Transition Metal Complexes. J. Phys. Chem. A 1997, 101, 2502–2508. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Q.; Zhou, L. Theoretical Studies on N–O or N–N Bond Formation from Aryl Azide Catalyzed by Iron(II) Bromide Complex. J. Org. Chem. 2012, 77, 2566–2570. [Google Scholar] [CrossRef]
- Check, C.E.; Faust, T.O.; Bailey, J.M.; Wright, B.J.; Gilbert, T.M.; Sunderlin, L.S. Addition of Polarization and Diffuse Functions to the LANL2DZ Basis Set for P-Block Elements. J. Phys. Chem. A 2001, 105, 8111–8116. [Google Scholar] [CrossRef]
- Li, Y.; Chen, H.; Qu, L.-B.; Houk, K.N.; Lan, Y. Origin of Regiochemical Control in Rh(III)/Rh(V)-Catalyzed Reactions of Unsaturated Oximes and Alkenes to Form Pyrdines. ACS Catal. 2019, 9, 7154–7165. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Addicoat, M.A.; Vankova, N.; Akter, I.F.; Heine, T. Extension of the Universal Force Field to Metal−Organic Frameworks. J. Chem. Theory Comput. 2014, 10, 880–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Anoop, A.; Thiel, W.; Neese, F.A. Local Pair Natural Orbital Coupled Cluster Study of Rh Catalyzed Asymmetric Olefin Hydrogenation. J. Chem. Theory Comput. 2010, 6, 3137–3144. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Meng, X.; Hu, C.; Su, Z. Cooperative Catalysis of Chiral Guanidine and Rh2(OAc)4 in Asymmetric O–H Insertion of Carboxylic Acid: A Theoretical Investigation. J. Org. Chem. 2019, 84, 15020–15031. [Google Scholar] [CrossRef]
- Legault, C.Y. CYLview; 1.0b; Université de Sherbrooke: Sherbrooke, QC, Canada, 2009; Available online: http://www.cylview.org (accessed on 31 October 2020).
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Henon, E. Accurately Extracting the Signature of Intermolecular Interactions Present in the NCI Plot of the Reduced Density Gradient Versus Electron Density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Lin, J.-S.; Li, T.-T.; Liu, J.-R.; Jiao, G.-Y.; Gu, Q.-S.; Cheng, J.-T.; Guo, Y.-L.; Hong, X.; Liu, X.-Y. Cu/Chiral Phosphoric Acid-Catalyzed Asymmetric Three-Component Radical-Initiated 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc. 2019, 141, 1074–1083. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. A New Local Density Functional for Main-Group Thermochemistry, Transition Metal Bonding, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Phys. 2006, 125, 194101–194118. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.S.; He, X.; Li, S.L.; Truhlar, D.G. MN15: A Kohn–Sham Global-Hybrid Exchange–Correlation Density Functional with Broad Accuracy for Multi-Reference and Single-Reference Systems and Noncovalent Interactions. Chem. Sci. 2016, 7, 5032–5051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, X.-X.; Chen, X.-H.; Ding, D.-B.; She, Y.-B.; Yang, Y.-F. Computational Exploration of Dirhodium Complex-Catalyzed Selective Intermolecular Amination of Tertiary vs. Benzylic C−H Bonds. Molecules 2023, 28, 1928. https://doi.org/10.3390/molecules28041928
Su X-X, Chen X-H, Ding D-B, She Y-B, Yang Y-F. Computational Exploration of Dirhodium Complex-Catalyzed Selective Intermolecular Amination of Tertiary vs. Benzylic C−H Bonds. Molecules. 2023; 28(4):1928. https://doi.org/10.3390/molecules28041928
Chicago/Turabian StyleSu, Xing-Xing, Xia-He Chen, De-Bo Ding, Yuan-Bin She, and Yun-Fang Yang. 2023. "Computational Exploration of Dirhodium Complex-Catalyzed Selective Intermolecular Amination of Tertiary vs. Benzylic C−H Bonds" Molecules 28, no. 4: 1928. https://doi.org/10.3390/molecules28041928