
Catalytic Oxidation of Benzoins by Hydrogen Peroxide on Nanosized HKUST-1: Influence of Substituents on the Reaction Rates and DFT Modeling of the Reaction Path

 , and
, and
Abstract
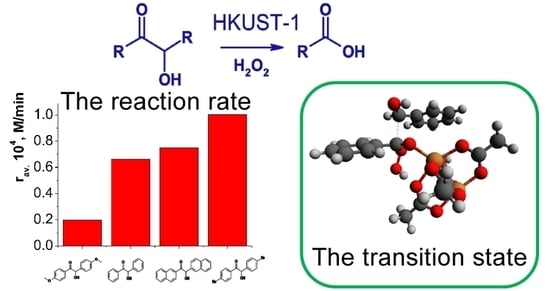
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization of Nanosized HKUST-1
2.2. Catalytic Oxidation of Benzoin and Analogs by H2O2
2.3. Analysis and DFT Calculations of the Reaction Pathway
3. Experimental and Computational Details
3.1. Materials and Methods
3.2. Synthesis of HKUST-1
3.3. Oxidation of Benzoin and Substituted Benzoins
3.4. Studies of the Adsorption of Reagents on HKUST-1
3.5. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Purtsas, A.; Rosenkranz, M.; Dmitrieva, E.; Kataeva, O.; Knölker, H.-J. Iron-Catalyzed Oxidative C−O and C−N Coupling Reactions Using Air as Sole Oxidant. Chem. Eur. J. 2022, 28, e202104292. [Google Scholar] [CrossRef] [PubMed]
- Senthamarai, T.; Chandrashekhar, V.G.; Rockstroh, N.; Rabeah, J.; Bartling, S.; Jagadeesh, R.V.; Beller, M. A “universal” catalyst for aerobic oxidations to synthesize (hetero)aromatic aldehydes, ketones, esters, acids, nitriles, and amides. Chem 2022, 8, 508–531. [Google Scholar] [CrossRef]
- Mallat, T.; Baiker, A. Oxidation of Alcohols with Molecular Oxygen on Solid Catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Fang, R.; Luque, R.; Chen, L.; Li, Y. Functional Metal-Organic Frameworks for Catalytic Applications. Coord. Chem. Rev. 2019, 388, 268–292. [Google Scholar] [CrossRef]
- Gangu, K.K.; Jonnalagadda, S.B. A Review on Metal-Organic Frameworks as Congenial Heterogeneous Catalysts for Potential Organic Transformations. Front. Chem. 2021, 9, 747615. [Google Scholar] [CrossRef]
- Sha, O.; Chuan-De, W. Rational Construction of Metal–Organic Frameworks for Heterogeneous Catalysis. Inorg. Chem. Front. 2014, 1, 721–734. [Google Scholar] [CrossRef]
- Zhao, M.; Yuan, K.; Wang, Y.; Li, G.; Guo, J.; Gu, L.; Hu, W.; Zhao, H.; Tang, Z. Metal-organic Frameworks as Selectivity Regulators for Hydrogenation Reactions. Nature 2016, 539, 76–80. [Google Scholar] [CrossRef]
- Guo, J.; Qin, Y.; Zhu, Y.; Zhang, X.; Long, C.; Zhao, M.; Tang, X. Metal–organic frameworks as catalytic selectivity regulators for organic transformations. Chem. Soc. Rev. 2021, 50, 5366–5396. [Google Scholar] [CrossRef]
- Kang, Y.-S.; Lu, Y.; Chen, K.; Zhao, Y.; Wang, P.; Sun, W.-Y. Metal-Organic Frameworks with Catalytic Centers: From Synthesis to Catalytic Application. Coord. Chem. Rev. 2019, 378, 262–280. [Google Scholar] [CrossRef]
- Aguirre-Díaz, L.M.; Gándara, F.; Iglesias, M.; Snejko, N.; Gutiérrez-Puebla, E.; Monge, M.Á. Tunable Catalytic Activity of Solid Solution Metal-Organic Frameworks in One-Pot Multicomponent Reactions. J. Am. Chem. Soc. 2015, 137, 6132–6135. [Google Scholar] [CrossRef]
- Dybtsev, D.N.; Bryliakov, K.P. Asymmetric catalysis using metal-organic frameworks. Coord. Chem. Rev. 2021, 437, 213845. [Google Scholar] [CrossRef]
- Satska, Y.A.; Komarova, N.P.; Gavrilenko, K.S.; Manoylenko, O.V.; Chernenko, Z.V.; Kiskin, M.A.; Kolotilov, S.V.; Eremenko, I.L.; Novotortsev, V.M. Sorption and Separation of Optical Isomers of 2-Butanol by Chiral Porous Coordination Polymers. Theor. Exp. Chem. 2015, 51, 45–53. [Google Scholar] [CrossRef]
- Zhai, Z.-W.; Yang, S.-H.; Lv, Y.-R.; Du, C.-X.; Li, L.-K.; Zang, S.-Q. Amino Functionalized Zn/Cd-Metal-Organic Frameworks for Selective CO2 Adsorption and Knoevenagel Condensation Reactions. Dalton Trans. 2019, 48, 4007–4014. [Google Scholar] [CrossRef]
- Sotnik, S.A.; Polunin, R.A.; Kiskin, M.A.; Kirillov, A.M.; Dorofeeva, V.N.; Gavrilenko, K.S.; Eremenko, I.L.; Novotortsev, V.M.; Kolotilov, S.V. Heterometallic Coordination Polymers Assembled from Trigonal Trinuclear Fe2Ni-Pivalate Blocks and Polypyridine Spacers: Topological Diversity, Sorption, and Catalytic Properties. Inorg. Chem. 2015, 54, 5169–5181. [Google Scholar] [CrossRef]
- Gong, W.; Liu, Y.; Li, H.; Cui, Y. Metal-organic Frameworks as Solid Brønsted Acid Catalysts for Advanced Organic Transformations. Coord. Chem. Rev. 2020, 420, 213400. [Google Scholar] [CrossRef]
- Hu, Z.; Zhao, D. Metal-Organic Frameworks with Lewis Acidity: Synthesis, Characterization, and Catalytic Applications. CrystEngComm 2017, 19, 4066–4081. [Google Scholar] [CrossRef]
- Lytvynenko, A.S.; Kolotilov, S.V. Electrochemically active coordination polymers: A review. Theor. Exp. Chem. 2016, 52, 197–211. [Google Scholar] [CrossRef]
- Lytvynenko, A.S.; Kolotilov, S.V.; Kiskin, M.A.; Cador, O.; Golhen, S.; Aleksandrov, G.G.; Mishura, A.M.; Titov, V.E.; Ouahab, L.; Eremenko, I.L.; et al. Redox-Active porous coordination polymers prepared by trinuclear heterometallic pivalate linking with the redox-active nickel(II) complex: Synthesis, structure, magnetic and redox properties, and electrocatalytic activity in organic compound dehalogenation in heterogeneous medium. Inorg. Chem. 2014, 53, 4970–4979. [Google Scholar] [CrossRef]
- Lytvynenko, A.S.; Kiskin, M.A.; Dorofeeva, V.N.; Mishura, A.M.; Titov, V.E.; Kolotilov, S.V.; Eremenko, I.L.; Novotortsev, V.M. Redox-active porous coordination polymer based on trinuclear pivalate: Temperature-dependent crystal rearrangement and redox-behavior. J. Solid State Chem. 2015, 223, 122–130. [Google Scholar] [CrossRef]
- Bukowski, B.C.; Snurr, R.Q. Topology-Dependent Alkane Diffusion in Zirconium Metal–Organic Frameworks. ACS Appl. Mater. Interfaces 2020, 12, 56049–56059. [Google Scholar] [CrossRef]
- Sotnik, S.A.; Gavrilenko, K.S.; Lytvynenko, A.S.; Kolotilov, S.V. Catalytic activity of copper (II) benzenetricarboxylate (HKUST-1) in reactions of aromatic aldehydes condensation with nitromethane: Kinetic and diffusion study. Inorg. Chim. Acta 2015, 426, 119–125. [Google Scholar] [CrossRef]
- Sikdar, A.; Majumdar, A.; Gogoi, A.; Dutta, P.; Borah, M.; Maiti, S.; Gogoi, C.; Reddy, K.A.; Oh, Y.; Maiti, U.N. Diffusion driven nanostructuring of metal–organic frameworks (MOFs) for graphene hydrogel based tunable heterostructures: Highly active electrocatalysts for efficient water oxidation. J. Mater. Chem. A 2021, 9, 7640–7649. [Google Scholar] [CrossRef]
- Cai, X.; Xie, Z.; Li, D.; Kassymova, M.; Zang, S.-Q.; Jiang, H.-L. Nano-sized metal-organic frameworks: Synthesis and applications. Coord. Chem. Rev. 2020, 417, 213366. [Google Scholar] [CrossRef]
- Ahmed, I.; Jhung, S.H. Composites of metal–organic frameworks: Preparation and application in adsorption. Mater. Today 2014, 17, 136–146. [Google Scholar] [CrossRef]
- Chen, L.; Xu, Q. Metal-Organic Framework Composites for Catalysis. Matter 2019, 1, 57–89. [Google Scholar] [CrossRef] [Green Version]
- Peng, Z.; Jimenez, J.L. Radical chemistry in oxidation flow reactors for atmospheric chemistry research. Chem. Soc. Rev. 2020, 49, 2570–2616. [Google Scholar] [CrossRef]
- Chui, S.S.Y.; Lo, S.M.F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef]
- Prestipino, C.; Regli, L.; Vitillo, J.G.; Bonino, F.; Damin, A.; Lamberti, C.; Zecchina, A.; Solari, P.L.; Kongshaug, K.O.; Bordiga, S. Local Structure of Framework Cu(II) in HKUST-1 Metallorganic Framework: Spectroscopic Characterization upon Activation and Interaction with Adsorbates. Chem. Mater. 2006, 18, 1337–1346. [Google Scholar] [CrossRef]
- Yepez, R.; García, S.; Schachat, P.; Sánchez-Sánchez, M.; González-Estefan, J.H.; González-Zamora, E.; Ibarra, I.A. Aguilar-Pliego, Catalytic activity of HKUST-1 in the oxidation of trans-ferulic acid to vanillin. New J. Chem. 2015, 39, 5112–5115. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, B.; Zhou, Q.; Zhang, T.; Wu, W. Morphology Effect of Metal-organic Framework HKUST-1 as a Catalyst on Benzene Oxidation. Chem. Res. Chin. Univ. 2017, 33, 971–978. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Metal organic frameworks as efficient heterogeneous catalysts for the oxidation of benzylic compounds with t-butylhydroperoxide. J. Catal. 2009, 267, 1–4. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Alvaro, M.; Garcia, H. Aerobic Oxidation of Benzylic Alcohols Catalyzed by Metal-Organic Frameworks Assisted by TEMPO. ACS Catal. 2011, 1, 48–53. [Google Scholar] [CrossRef]
- Lei, M.; Hu, R.-J.; Wang, Y.-G. Mild and selective oxidation of alcohols to aldehydes and ketones using NaIO4/TEMPO/NaBr system under acidic conditions. Tetrahedron 2006, 62, 8928–8932. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, J.; Ma, S. Iron Catalysis for Room-Temperature Aerobic Oxidation of Alcohols to Carboxylic Acids. J. Am. Chem. Soc. 2016, 138, 8344–8347. [Google Scholar] [CrossRef]
- Yan, J.; Travis, B.R.; Borhan, B. Direct Oxidative Cleavage of α- and β-Dicarbonyls and α-Hydroxyketones to Diesters with KHSO5. J. Org. Chem. 2004, 69, 9299–9302. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cao, R.; Lin, Q. Solvent-free Baeyer–Villiger oxidation with H2O2 as oxidant catalyzed by multi-SO3H functionalized heteropolyanion-based ionic hybrids. Catal. Commun. 2015, 63, 79–83. [Google Scholar] [CrossRef]
- Lei, Z.; Ma, G.; Wei, L.; Yang, Q.; Su., B. Clean Baeyer–Villiger Oxidation Using Hydrogen Peroxide as Oxidant Catalyzed by Aluminium Trichloride in Ethanol. Catal Lett. 2008, 124, 330–333. [Google Scholar] [CrossRef]
- Lei, Z.; Zhang, Q.; Wang, R.; Ma, G.; Jia, C. Clean and selective Baeyer–Villiger oxidation of ketones with hydrogen peroxide catalyzed by Sn-palygorskite. J. Organomet. Chem. 2006, 691, 5767–5773. [Google Scholar] [CrossRef]
- Chen, S.-Y.; Zhou, X.-T.; Ji, H.-B. Insight into the cocatalyst effect of 4A molecular sieve on Sn(II) porphyrin-catalyzed B–V oxidation of cyclohexanone. Catal. Today 2016, 264, 191–197. [Google Scholar] [CrossRef]
- Strukul, G. Transition Metal Catalysis in the Baeyer-Villiger Oxidation of Ketones. Angew. Chem. Int. Ed. 1998, 37, 1198–1209. [Google Scholar] [CrossRef]
- Gorban, O.; Danilenko, I.; Nosolev, I.; Abdullayev, E.; Islamov, A.; Gavrilenko, K.; Doroshkevich, A.; Shvets, O.; Kolotilov, S. Impact of chemical and physical modification of zirconia on structure, surface state, and catalytic activity in oxidation of α-tetralol. J. Nanopart. Res. 2022, 24, 197. [Google Scholar] [CrossRef]
- Münch, A.S.; Mertens, F.O.R.L. The Lewis acidic and basic character of the internal HKUST-1 surface determined by inverse gas chromatography. CrystEngComm 2015, 17, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Borfecchia, E.; Maurelli, S.; Gianolio, D.; Groppo, E.; Chiesa, M.; Bonino, F.; Lamberti, C. Insights into Adsorption of NH3 on HKUST-1 Metal–Organic Framework: A Multitechnique Approach. J. Phys. Chem. C 2012, 116, 19839–19850. [Google Scholar] [CrossRef]
- Fuson, R.S.; Horning, E.C. Enediols. V. Hexaisopropylstilbenediols. J. Am. Chem. Soc. 1940, 62, 2962–2964. [Google Scholar] [CrossRef]
- Weiβberger, A.; Mainz, H.; Strasser, E. Über die Autoxydation des Benzoins in alkalischer Lösung. Berichte Dtsch. Chem. Ges. (A B Ser.) 1929, 62, 1942–1952. [Google Scholar] [CrossRef]
- Weitz, E.; Scheffer, A. Umwandlungen der Ketoxidoverbindungen; Bildung von β-Keto-aldehyden aus α,β-ungesättigten Ketonen. Ber. Dtsch. Chem. Ges. (A B Ser.) 1921, 54, 2344–2353. [Google Scholar] [CrossRef]
- Duc, D.X.; Nguyet, N.T. Copper triflate catalyzed Baeyer-Villiger oxidation of ketones. Vietnam J. Sci. Technol. 2019, 57, 76–81. [Google Scholar] [CrossRef]
- Bolm, C.; Schlingloff, G.; Weickhardt, K. Optically Active Lactones from a Baeyer-Villiger-Type Metal-Catalyzed Oxidation with Molecular Oxygen. Angew. Chem. Int. Ed. 1994, 33, 1848–1849. [Google Scholar] [CrossRef]
- Jiao, N.; Stahl, S.S. (Eds.) Green Oxidation in Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar] [CrossRef]
- Młochowski, J.; Peczyńska-Czoch, W.; Piętka-Ottlik, M.; Wójtowicz-Młochowska, H. Non-Metal and Enzymatic Catalysts for Hydroperoxide Oxidation of Organic Compounds. Open Catal. J. 2011, 4, 54–82. [Google Scholar] [CrossRef]
- Shul’pin, G.B.; Shul’pina, L.S. Oxidation of Organic Compounds with Peroxides Catalyzed by Polynuclear Metal Compounds. Catalysts 2021, 11, 186. [Google Scholar] [CrossRef]
- Miller, C.J.; Rose, A.L.; Waite, T.D. Hydroxyl Radical Production by H2O2-Mediated Oxidation of Fe(II) Complexed by Suwannee River Fulvic Acid Under Circumneutral Freshwater Conditions. Environ. Sci. Technol. 2013, 47, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Florence, T.M. The production of hydroxyl radical from hydrogen peroxide. J. Inorg. Biochem. 1984, 22, 221–230. [Google Scholar] [CrossRef]
- Joaristi, A.M.; Juan-Alcañiz, J.; Serra-Crespo, P.; Kapteijn, F.; Gascon, J. Electrochemical Synthesis of Some Archetypical Zn2+, Cu2+, and Al3+ Metal Organic Frameworks. Cryst. Growth Des 2012, 12, 3489–3498. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0-new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Patterson, A.L. The Scherrer formula for X-ray particle size determination. Phys. Rev. 1939, 56, 978–982. [Google Scholar] [CrossRef]
- Chowdhury, P.; Bikkina, C.; Meister, D.; Dreisbach, F.; Gumma, S. Comparison of adsorption isotherms on Cu-BTC metal organic frameworks synthesized from different routes. Microporous Mesoporous Mater. 2009, 117, 406–413. [Google Scholar] [CrossRef]
- Maurya, M.R.; Arya, A.; Kumar, A.; Pessoa, J.C. Polystyrene bound oxidovanadium(IV) and dioxidovanadium(V) complexes of histamine derived ligand for the oxidation of methyl phenyl sulfide, diphenyl sulfide and benzoin. Dalton Trans. 2009, 12, 2185–2195. [Google Scholar] [CrossRef]
- Maurya, M.R.; Dhaka, S.; Avecilla, F. Synthesis, characterization and catalytic activity of dioxidomolybdenum(VI) complexes of tribasic pentadentate ligands. Polyhedron 2013, 67, 145–159. [Google Scholar] [CrossRef]
- Maurya, M.R.; Chaudhary, N.; Kumar, A.; Avecilla, F.; Pessoa, J.C. Polystyrene bound dioxidovanadium(V) complexes of 2-acetylpyridine derived ligands for catalytic oxidations. Inorg. Chim. Acta 2013, 420, 24–38. [Google Scholar] [CrossRef]
- Pasayat, S.; Dash, S.P.; Roy, S.; Dinda, R.; Dhaka, S.; Maurya, M.R.; Kaminsky, W.; Patil, Y.P.; Nethaji, M. Synthesis, structural studies and catalytic activity of dioxidomolybdenum(VI) complexes with aroylhydrazones of naphthol-derivative. Polyhedron 2014, 67, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Maurya, M.R.; Saini, P.; Haldar, C.; Avecilla, F. Synthesis, characterisation and catalytic activities of manganese(III) complexes of pyridoxal-based ONNO donor tetradenatate ligands. Polyhedron 2012, 31, 710–720. [Google Scholar] [CrossRef]
- Ho, Y.S.; McKay, G. Pseudo-second order model for sorption processes. Process Biochem. 1999, 34, 451–465. [Google Scholar] [CrossRef]
- Bunton, C.A. Oxidation of α-Diketones and α-Keto-acids by Hydrogen Peroxide. Nature 1949, 163, 444. [Google Scholar] [CrossRef]
- Kremer, M.L. Strong inhibition of the Fe3+ + H2O2 reaction by ethanol: Evidence against the free radical theory. Prog. React. Kinet. Mech. 2017, 42, 397–413. [Google Scholar] [CrossRef]
- Tariq, S.; Khalid, M.; Raza, A.R.; Rubab, S.L.; Figueirêdo de Alcântara Morais, S.; Khan, M.U.; Tahir, M.N.; Braga, A.A.C. Experimental and computational investigations of new indole derivatives: A combined spectroscopic, SC-XRD, DFT/TD-DFT and QTAIM analysis. J. Mol. Struct. 2020, 1207, 127803. [Google Scholar] [CrossRef]
- Khan, E.; Khalid, M.; Gul, Z.; Shahzad, A.; Tahir, M.N.; Asif, H.M.; Asim, S.; Braga, A.A.C. Molecular structure of 1,4-bis(substituted-carbonyl)benzene: A combined experimental and theoretical approach. J. Mol. Struct. 2020, 1205, 127633. [Google Scholar] [CrossRef]
- Tariq, S.; Raza, A.R.; Khalid, M.; Rubab, S.L.; Khan, M.U.; Ali, A.; Tahir, M.N.; Braga, A.A.C. Synthesis and structural analysis of novel indole derivatives by XRD, spectroscopic and DFT studies. J. Mol. Struct. 2020, 1203, 127438. [Google Scholar] [CrossRef]
- Lytvynenko, A.S.; Kolotilov, S.V.; Kiskin, M.A.; Eremenko, I.L.; Novotortsev, V.M. Modeling of catalytically active metal complex species and intermediates in reactions of organic halides electroreduction. Phys. Chem. Chem. Phys. 2015, 17, 5594–5605. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent Radii Revisited. Dalton Trans. 2008, 21, 2832–2838. [Google Scholar] [CrossRef]
- Murzin, D.Y.; Bertrand, E.; Tolvanen, P.; Devyatkov, S.; Rahkila, J.; Eränen, K.; Wärnå, J.; Salmi, T. Heterogeneous Catalytic Oxidation of Furfural with Hydrogen Peroxide over Sulfated Zirconia. Ind. Eng. Chem. Res. 2020, 59, 13516–13527. [Google Scholar] [CrossRef]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Adams, R.; Marvel, C.S. Benzoin. Org. Synth. 1921, 1, 33. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Neese, F. Software Update: The ORCA Program System, Version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA Quantum Chemistry Program Package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully Optimized Contracted Gaussian Basis Sets for Atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571. [Google Scholar] [CrossRef] [Green Version]
- Weigend, F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868, Erratum in Phys. Rev. Lett. 1997, 78, 1396. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Barone, V.; Cossi, M. Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A 1998, 102, 1995–2001. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a Full Periodic Table Force Field for Molecular Mechanics and Molecular Dynamics Simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, G.; Jónsson, H.; Schenter, G.K. Reversible Work Transition State Theory: Application to Dissociative Adsorption of Hydrogen. Surf. Sci. 1995, 324, 305–337. [Google Scholar] [CrossRef] [Green Version]
- Henkelman, G.; Jónsson, H. Improved Tangent Estimate in the Nudged Elastic Band Method for Finding Minimum Energy Paths and Saddle Points. J. Chem. Phys. 2000, 113, 9978–9985. [Google Scholar] [CrossRef] [Green Version]
- Ásgeirsson, V.; Birgisson, B.O.; Bjornsson, R.; Becker, U.; Neese, F.; Riplinger, C.; Jónsson, H. Nudged Elastic Band Method for Molecular Reactions Using Energy-Weighted Springs Combined with Eigenvector Following. J. Chem. Theory Comput. 2021, 17, 4929–4945. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Atomic Dipole Moment Corrected Hirshfeld Population Method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining Atom-Centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Hirshfeld, F.L. Bonded-Atom Fragments for Describing Molecular Charge Densities. Theor. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Becke, A.D. A Multicenter Numerical Integration Scheme for Polyatomic Molecules. J. Chem. Phys. 1988, 88, 2547–2553. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]


















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yurchenko, D.V.; Lytvynenko, A.S.; Abdullayev, E.N.; Peregon, N.V.; Gavrilenko, K.S.; Gorlova, A.O.; Ryabukhin, S.V.; Volochnyuk, D.M.; Kolotilov, S.V. Catalytic Oxidation of Benzoins by Hydrogen Peroxide on Nanosized HKUST-1: Influence of Substituents on the Reaction Rates and DFT Modeling of the Reaction Path. Molecules 2023, 28, 747. https://doi.org/10.3390/molecules28020747
Yurchenko DV, Lytvynenko AS, Abdullayev EN, Peregon NV, Gavrilenko KS, Gorlova AO, Ryabukhin SV, Volochnyuk DM, Kolotilov SV. Catalytic Oxidation of Benzoins by Hydrogen Peroxide on Nanosized HKUST-1: Influence of Substituents on the Reaction Rates and DFT Modeling of the Reaction Path. Molecules. 2023; 28(2):747. https://doi.org/10.3390/molecules28020747
Chicago/Turabian StyleYurchenko, Darya V., Anton S. Lytvynenko, Emir N. Abdullayev, Nina V. Peregon, Konstantin S. Gavrilenko, Alina O. Gorlova, Sergey V. Ryabukhin, Dmitriy M. Volochnyuk, and Sergey V. Kolotilov. 2023. "Catalytic Oxidation of Benzoins by Hydrogen Peroxide on Nanosized HKUST-1: Influence of Substituents on the Reaction Rates and DFT Modeling of the Reaction Path" Molecules 28, no. 2: 747. https://doi.org/10.3390/molecules28020747