
Quality Control of Pesticide Residue Measurements and Evaluation of Their Results

 ,
,
Abstract
:1. Introduction
2. Methods
2.1. Sampling
2.2. Selection of Portion of Sample to Be Analysed
2.3. Subsampling and Comminution of Selected Sample Portions
2.4. Definition of Residues
2.5. Extraction of Residues and Cleanup of Extracts
2.6. Accuracy of Standard Solutions
2.7. Stability of Analytes
- (a)
- Take about two or more kg of the crop in which the stability of analytes will be tested.
- (b)
- Prepare the portion of the commodity to be analyzed according to Codex CAC/GL 41-1993 [81] from the whole laboratory sample.
- (c)
- Use approximately half of the sample matrix for the stability test and the remaining part for the recovery tests performed with spiked test portions as usual.
- (d)
- Prepare analytical standard mixture of exactly known concentration of compounds to be tested together with buprofezin (Bu) and chlorpyrifos (Ch) at well detectable concentrations keeping in mind the total mass of the sub-sample to be processed. The number of pesticides or metabolites included in the mixture is limited only by the capability of the chromatographic separation and detection system.
- (e)
- Take about 1/3 of the part of the laboratory sample (e.g., 3–4 units out of 10 fruits) obtained in step 3;
- (f)
- Treat the surface of the selected portion applying either Hamilton syringe for carefully spreading the standard mixture (step 4) on the surface of the crops or injecting the standard solution into the flesh of the fruit [106]. Use liquid dispenser to treat leafy vegetables or small-size crops. Perform the treatment in a fume cupboard over a tray with filter paper which can absorb the runoff. The exact amount of standard mixture that remains on the crop surface need not be known as the concentration ratios of the reference and test compounds will be calculated.
- (g)
- Steps for the treatment of the surface of tomatoes:
- Place the surface-treated portions into the chopper together with the remaining 2/3 part of the sample and comminute the whole matrix. By this way you represent a potentially worst-case scenario for testing the efficiency of sample processing and determination of CVL at the same time as testing the stability of analytes. The test may be performed both at ambient temperature and under cryogenic conditions applying dry ice or liquid nitrogen following the normal procedure applied in the laboratory.
- Verify the efficiency of comminution with a Petri dish test (See Section 2.3). Continue the process until an acceptable particle size distribution is obtained. Note that a lengthy process may increase the decomposition.
- Remove test portions from the comminuted matrix according to the normal procedure of the laboratory, but preferably from≥ 10 different positions.
- (h)
- Using the remaining part of the test material, perform the recovery test as usual by spiking the selected test portion with the standard mixture. Calculate the recovery for each compound.
- (i)
- Determine the concentration of survived residues from the surface-treated material and their recovery from spike test portions with the method to be applied.
- (j)
- Perform the test in ≥5 replicates.
Calculation of the Stability of Test Compounds
2.8. Determination and Demonstration of within-Laboratory Reproducibility
2.9. Chromatographic Determination of Residues


3. Discussion
- dietary exposure assessment of consumers;
- evaluating the residue levels and their compliance with national or international maximum residue limits or guidance values;
- assessing the contamination of the environment;
- providing the basis for the necessary corrective actions if the residues exceed the reasonably expectable levels in the treated crops.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| APVMA | Australian Pesticides and Veterinary Medicines Authority |
| EPA | US Environmental Protection Agency |
| ADI | Acceptable Daily Intake |
| ANVISA | Brazil National Health Surveillance Agency |
| CAC | Codex Alimentarius Commission |
| CCPR | Codex Committee on Pesticide Residues |
| DAFF | Australian Government Department of Agriculture, Fisheries and Forestry |
| EC | European Commission |
| EFSA | European Food Safety Authority |
| EPC | European Parliament and Council |
| EU | European Union |
| FAO | Food and Agriculture Organization of the United Nations |
| FIFRA | Federal Insecticide, Fungicide and Rodenticide Act |
| GAP | Good Agricultural Practice |
| JMH | Ministry of Health of Japan |
| JMPR | FAO/WHO Joint Meeting on Pesticide Residues |
| MRL | Maximum Residue Limit [mg/kg] |
| OECD | Organisation for Economic Cooperation and Development |
| USA | United States of America |
| US EPA | US Environmental Protection Agency |
| US FDA | US Food and Drug Administration |
References
- FAO/WHO. Synthesis Report on the Environmental and Health Impacts of Pesticides and Fertilizers and Ways to Minimize Them. 2022. Available online: www.fao.org/pest-and-pesticide-management/resources/en/ (accessed on 9 November 2022).
- National Research Council, 2020 Future Role of Pesticides in US Agriculture; National Academies Press: Washington, DC, USA, 2020; p. 332. ISBN 0-309-50383-3.
- Turner, A. Pesticide Manual, 19th ed.British Crop Production Council: Alton, UK, 2021; p. 1400. ISBN 978-1-9998966-5-2. [Google Scholar]
- University of Hertfordshire (Hatfield, UK). Pesticide Property Database. Available online: http://sitem.herts.ac.uk/aeru/ppdb/en/Reports/3478.htm (accessed on 20 October 2022).
- EFSA. Eurobarometer. 2022. Available online: https://www.efsa.europa.eu/sites/default/files/2022-09/EB97.2-food-safety-in-the-EU_report.pdf (accessed on 24 November 2022).
- Australian Government. Agricultural and veterinary chemicals code, standards. 2022. Available online: https://www.legislation.gov.au/Details/F2022L00137 (accessed on 20 October 2022).
- European Parliament and Council. Regulation (EC) no. 396/2005 of the European Parliament and of the Council of 23 February 2005 on Maximum Residue Levels of Pesticides in or on Food and Feed of Plant and Animal Origin and Amending Council Directive 91/414/EEC. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=celex%3A32005R0396 (accessed on 20 October 2022).
- European Parliament and Council. Regulation (EC) no. 1107/2009 of the European Parliament and of the Council of 21 October 2009 Concerning the Placing of Plant Protection Products on the Market and Repealing Council Directives 79/117/EEC and 91/414/EEC. Available online: https://eur-lex.europa.eu/legal-content/HU/ALL/?uri=CELEX:32009R1107 (accessed on 20 October 2022).
- EPA. Summary of the Federal Insecticide, Fungicide and Rodenticide Act. Available online: https://www.epa.gov/laws-regulations/summary-federal-insecticide-fungicide-and-rodenticide-act (accessed on 20 October 2022).
- EPA. Regulation of Pesticide Residues on Food. 2022. Available online: https://www.epa.gov/pesticide-tolerances (accessed on 5 November 2022).
- Japanese Ministry of Health, Labour and Welfare. Agricultural Chemical Residues in Foods. Available online: https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/shokuhin/zanryu/index_00016.htm (accessed on 5 November 2022).
- Solecki, R.; Schumacher, D.M.; Pfeil, R.; Bhula, R. OECD Guidance documents and test guidelines. In Food Safety Assessment of Pesticide Residues; Ambrus, Á., Hamilton, D., Eds.; World Scientific: Hackensack, NJ, USA, 2017; pp. 507–510. [Google Scholar]
- OECD. Test Guidelines for Chemicals. Available online: https://www.oecd.org/chemicalsafety/testing/oecdguidelinesforthetestingofchemicals.htm (accessed on 5 November 2022).
- OECD. Work on Pesticides and Sustainable Pest Management. Vision for the Future. Available online: https://inergency.com/oecd-work-on-pesticides-and-sustainable-pest-management-vision-for-the-future (accessed on 5 November 2022).
- OECD. Data Requirements for Pesticide Registration in OECD Member Countries: Survey Results. OECD/GD (94)47 Environment Monographs No. 77. Available online: https://www.oecd.org/env/ehs/pesticides-biocides/oecdguidancedocumentsforpesticideregistration.htm (accessed on 5 November 2022).
- ASEAN. Maximum Residue Levels. Available online: https://asean.org/wp-content/uploads/images/2015/October/outreach-document/Edited%20MRLs-2.pdf (accessed on 5 November 2022).
- Brazil Government. Biblioteca de Temas de Agrotóxicos. 2022. Available online: https://www.gov.br/anvisa/pt-br/assuntos/regulamentacao/legislacao/bibliotecas-tematicas/arquivos/agrotoxicos.pdf (accessed on 5 November 2022).
- Vietnam. Ministry of Healthy Circular 50/2016 Regulations on MRL of Pesticide in Food. Available online: https://thuvienphapluat.vn/van-ban/The-thao-Y-te/Thong-tu-50-2016-TT-BYT-gioi-han-toi-da-du-luong-thuoc-bao-ve-thuc-vat-trong-thuc-pham-337490.aspx (accessed on 25 November 2022).
- Hamilton, D.; Yoshida, M.; Wolterink, G.; Solecki, R. Evaluation of pesticide residues by FAO/WHO JMPR. In Food Safety Assessment of Pesticide Residues, 1st ed.; Ambrus, Á., Hamilton, D., Eds.; World Scientific Publishing: London, UK, 2017; pp. 113–196. [Google Scholar]
- Yamada, Y. Importance of codex maximum residue limits for pesticides for the health of consumers and international trade. In Food Safety Assessment of Pesticide Residues, 1st ed.; Ambrus, Á., Hamilton, D., Eds.; World Scientific: Hackensack, NJ, USA, 2017; pp. 269–282. [Google Scholar]
- Codex Alimentarius. Pesticide Index. Available online: http://www.fao.org/fao-who-codexalimentarius/codex-texts/dbs/pestres/pesticides/en/ (accessed on 5 November 2022).
- Australian National Residue Survey. Available online: https://www.awe.gov.au/agriculture-land/farm-food-drought/food/nrs (accessed on 5 November 2022).
- Austrian Agency for Health and Food Safety. National Pesticide Residue Control Programmes. Available online: https://www.ages.at/en/plant/pesticides/pesticide-residues#c5252 (accessed on 30 October 2022).
- European Commission. Commission Implementing Regulation (EU) 2021/601 of 13 April 2021 Concerning a Coordinated Multiannual Control Programme of the Union for 2022, 2023 and 2024 to Ensure Compliance with Maximum Residue Levels of Pesticides and to Assess the Consumer Exposure to Pesticide Residues in and on Food of Plant and Animal Origin. Available online: http://data.europa.eu/eli/reg_impl/2021/601/oj (accessed on 22 October 2022).
- EFSA. The 2020 European Union Report on Pesticide Residues. Available online: https://multimedia.efsa.europa.eu/pesticides-report-2020/ (accessed on 5 November 2022).
- Federal Office of Consumer Protection, Tables for the National Reporting of Pesticide Residues in Food 2020. Available online: https://www.bvl.bund.de/DE/Arbeitsbereiche/01_Lebensmittel/01_Aufgaben/02_AmtlicheLebensmittelueberwachung/07_PSMRueckstaende/01_nb_psm_2020_tabellen/nbpsm_2020_tabellen_node.html (accessed on 20 October 2022).
- EPA 2015 Compliance Monitoring Strategy for Federal Insecticide, Fungicide, and Rodenticide Act (FIFRA)EC 2022a EU Pesticide Database. Available online: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/active-substances/?event=search.as (accessed on 20 September 2022).
- European Commission. EU Pesticide Database. Available online: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/active-substances/?event=search.as (accessed on 20 October 2022).
- USFDA. Pesticide Residue Monitoring Program Reports and Data. Available online: https://www.fda.gov/food/pesticides/pesticide-residue-monitoring-program-reports-and-data (accessed on 5 November 2022).
- Zhang, M.; Zeiss, M.R.; Geng, S. Agricultural pesticide use and food safety: California’s model. J. Integr. Agric. 2015, 14, 2340–2357. [Google Scholar] [CrossRef]
- Mac Loughlin, T.M.; Peluso, L.; Etchegoyen, A.; Alonso, L.L.; de Castro, C.; Percudani, C.; Marino, D.J.G. Pesticide residues in fruits and vegetables of the Argentine domestic market: Occurrence and quality. Food Control 2018, 93, 129–138. [Google Scholar] [CrossRef]
- Rahman, M.; Hoque, M.S.; Bhowmik, S.; Ferdousi, S.; Kabiraz, M.P.; van Brakel, M.L. Monitoring of pesticide residues from fish feed, fish and vegetables in Bangladesh by GC-MS using the QuEChERS method. Heliyon 2021, 7, e06390. [Google Scholar] [CrossRef] [PubMed]
- Jardim, A.N.O.; Caldas, E.D. Brazilian monitoring programs for pesticide residues in food-results from 2001 to 2010. Food Control 2012, 25, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Galani, Y.J.H.; Houbraken, M.; Wumbei, A.; Djeugap, J.F.; Fotio, D.; Gong, Y.Y.; Spanoghe, P. Monitoring and dietary risk assessment of 81 pesticide residues in 11 local agricultural products from the 3 largest cities of Cameroon. Food Control 2020, 118, 107416. [Google Scholar] [CrossRef]
- Calderon, R.; García-Hernández, J.; Palma, P.; Leyva-Morales, J.B.; Zambrano-Soria, M.; Bastidas-Bastidas, P.J.; Godoy, M. Assessment of pesticide residues in vegetables commonly consumed in Chile and Mexico: Potential impacts for public health. J. Food Compos. Anal. 2022, 108, 104420. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, M.; Silipunyo, T.; Huang, H.; Yin, Q.; Han, B.; Wang, M. Risk Assessment of Triflumezopyrim and Imidacloprid in rice through an evaluation of residual data. Molecules 2022, 27, 5685. [Google Scholar] [CrossRef]
- Gebremichael, S.; Birhanu, T.; Tessema, D.A. Analysis of organochlorine pesticide residues in human and cow’s milk in the towns of Asendabo, Serbo and Jimma in South-Western Ethiopia. Chemosphere 2013, 90, 1652–1657. [Google Scholar] [CrossRef]
- El Hawari, K.; Mokh, S.; Al Iskandarani, M.; Halloum, W.; Jaber, F. Pesticide residues in Lebanese apples and health risk assessment. Food Addit. Contam. B 2019, 12, 81–89. [Google Scholar] [CrossRef]
- Kottadiyil, D.; Mehta, T.; Thasale, R.; Sivaperumal, P. Determination and dietary risk assessment of 52 pesticide residues in vegetable and fruit samples by GC-MS/MS and UHPLC-QTOF/MS from Gujarat, India. J. Food Compos. Anal. 2023, 115, 104957. [Google Scholar] [CrossRef]
- Tripathy, V.; Sharma, K.K.; Sharma, K.; Gupta, R.; Yadav, R.; Singh, G.; Aggarwal, A.; Walia, S. Monitoring and dietary risk assessment of pesticide residues in brinjal, capsicum, tomato, and cucurbits grown in Northern and Western regions of India. J. Food Compos. Anal. 2022, 110, 104543. [Google Scholar] [CrossRef]
- Philippe, V.; Neveen, A.; Marwa, A.; Ahmad Basel, A.-Y. Occurrence of pesticide residues in fruits and vegetables for the Eastern Mediterranean Region and potential impact on public health. Food Control 2021, 119, 107457. [Google Scholar] [CrossRef]
- Ngabirano, H.; Birungi, G. Pesticide residues in vegetables produced in rural south-western Uganda. Food Chem. 2022, 370, 130972. [Google Scholar] [CrossRef] [PubMed]
- Osaili, T.M.; Al Sallagi, M.S.; Dhanasekaran, D.K.; Bani Odeh, W.A.M.; Al Ali, H.J.; Al Ali, A.A.S.A.; Radwan, H.; Obaid, R.S.; Holley, R. Pesticide residues in fresh vegetables imported into the United Arab Emirates. Food Control 2022, 133, 108663. [Google Scholar] [CrossRef]
- Ramadan, M.F.A.; Abdel-Hamid, M.M.A.; Altorgoman, M.M.F.; AlGaramah, H.A.; Alawi, M.A.; Shati, A.A.; Shweeta, H.A.; Awwad, N.S. Evaluation of pesticide residues in vegetables from the Asir Region Saudi Arabia. Molecules 2020, 25, 205. [Google Scholar] [CrossRef] [Green Version]
- Vu-Duc, N.; Nguyen-Quang, T.; Le-Minh, T.; Nguyen-Thi, X.; Tran, T.M.; Vu, H.A.; Chu, D.B. Multiresidue pesticides analysis of vegetables in Vietnam by ultrahigh-performance liquid chromatography in combination with high-resolution mass spectrometry (UPLC-Orbitrap MS). J. Anal. Methods Chem. 2019, 2019, 3489634. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Huynh Nguyen, K.; Duong, T.; Tran, V.D.; Kim, K.H.; Pham, L.T. Evaluation of pesticide residues in vegetables from Mekong, Delta, Vietnam using LC-MS/MS. Trop. J. Pharm. Res. 2021, 20, 1497–1504. [Google Scholar] [CrossRef]
- Carrasco Cabrera, L.; Medina Pastor, P. The 2020 European Union report on pesticide residues in food. EFSA J. 2022, 20, e07215. [Google Scholar] [CrossRef]
- Wu, P.; Wang, P.; Gu, M.; Xue, J.; Wu, X. Human health risk assessment of pesticide residues in honeysuckle samples from different planting bases in China. Sci. Total Environ. 2021, 759, 142747. [Google Scholar] [CrossRef]
- Pang, G.-F.; Fan, C.-L.; Liu, Y.-M.; Cao, Y.-Z.; Zhang, J.-J.; Fu, B.-L.; Li, X.-M.; Li, Z.Y.; Wu, Y.P. Multi-residue method for the determination of 450 pesticide residues in honey, fruit juice and wine by double-cartridge solid-phase extraction/gas chromatography-mass spectrometry and liquid chromatography-tandem mass spectrometry. Food Addit. Contam. 2006, 23, 777–810. [Google Scholar] [CrossRef]
- Pang, G.F.; Fan, C.L.; Chang, Q.Y.; Li, J.X.; Lu, J.K.L.M. Screening of 485 pesticide residues in fruits and vegetables by liquid chromatography-quadrupole-time-of-flight mass spectrometry based on TOF Accurate Mass Database and QTOF Spectrum Library. J. AOAC Int. 2018, 101, 1156–1182. [Google Scholar] [CrossRef] [PubMed]
- Tran-Lam, T.-T.; Bui, M.Q.; Nguyen, H.Q.; Dao, Y.H.; Le, G.T. A combination of chromatography with tandem mass spectrometry systems (UPLC-MS/MS and GC-MS/MS), modified QuEChERS extraction and mixed-mode SPE clean-up method for the analysis of 656 pesticide residues in rice. Foods 2021, 10, 2455. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and Easy Multiresidue Method Employing Acetonitrile Extraction/Partitioning and ‘dispersive solid-phase extraction’ for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Li, F.; Yan, T.; Tian, F.; Ren, L.; Jiang, C.; Wang, Q.; Zhang, S. Research progress in the sample pretreatment techniques and advanced quick detection methods of pesticide residues. Process Saf. Environ. Prot. 2022, 165, 610–622. [Google Scholar] [CrossRef]
- Calza, P.; Guarino, B.; Bello, F.D.; Dioni, A.; Bergero, M.; Medana, C. Integrated approach for the analysis of neonicotinoids in fruits and food matrices. Food Chem. 2022, 372, 131153. [Google Scholar] [CrossRef]
- Watanabe, E. Review of sample preparation methods for chromatographic analysis of neonicotinoids in agricultural and environmental matrices: From classical to state-of-the-art methods. J. Chromatogr. A 2021, 1643, 462042. [Google Scholar] [CrossRef]
- Rutkowska, E.; Łozowicka, B.; Kaczyński, P. Three approaches to minimize matrix effects in residue analysis of multiclass pesticides in dried complex matrices using gas chromatography tandem mass spectrometry. Food Chem. 2018, 279, 20–29. [Google Scholar] [CrossRef]
- Rutkowska, E.; Łozowicka, B.; Kaczyński, P. Compensation of matrix effects in seed matrices followed by gas chromatography-tandem mass spectrometry analysis of pesticide residues. J. Chromatogr. A 2019, 1614, 460738. [Google Scholar] [CrossRef]
- Musarurwa, H.; Chimuka, L.; Pakade, V.E.; Tavengwa, N.T. Recent developments and applications of QuEChERS based techniques on food samples during pesticide analysis. J. Food Compos. Anal. 2019, 84, 103314. [Google Scholar] [CrossRef]
- Xu, M.L.; Gao, Y.; Wang, X.; Han, X.X.; Zhao, B. Comprehensive strategy for sample preparation for the analysis of food contaminants and residues by GC–MS/MS: A review of recent research trends. Foods 2021, 10, 2473. [Google Scholar] [CrossRef]
- García-Vara, M.; Postigo, C.; Palma, P.; José Bleda, M.; López de Alda, M. QuEChERS-based analytical methods developed for LC-MS/MS multiresidue determination of pesticides in representative crop fatty matrices: Olives and sunflower seeds. Food Chem. 2022, 386, 132558. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, S.; Feng, H.; Yang, Y.; Xiao, B.; Chen, D. An enhanced sensitivity and cleanup strategy for the nontargeted screening and targeted determination of pesticides in tea using modified dispersive solid-phase extraction and cold-induced acetonitrile aqueous two-phase systems coupled with liquid chromatography-high resolution mass spectrometry. Food Chem. 2019, 275, 530–538. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed 11312/2021. Available online: https://food.ec.europa.eu/system/files/2022-02/pesticides_mrl_guidelines_wrkdoc_2021-11312.pdf (accessed on 5 November 2022).
- European Commission. Guidance Document on Pesticide Analytical Methods for Risk Assessment and Post-Approval Control and Monitoring Purposes SANTE/2020/12830, Rev. 1. Available online: https://food.ec.europa.eu/system/files/2021-02/pesticides_mrl_guidelines_2020-12830.pdf (accessed on 5 November 2022).
- US FDA. Guidelines for the Validation of Chemical Methods in Food, Feed, Cosmetics, and Veterinary Products, 3rd ed.; Food and Drug Administration: Silver Spring, MD, USA, 2020. Available online: https://www.fda.gov/media/121751/download (accessed on 5 November 2022).
- Ambrus, A. Reliability of measurements of pesticide residues in food. Accredit. Qual. Assur. 2004, 9, 288–304. [Google Scholar] [CrossRef]
- Ambrus, Á.; Zentai, A.; Sali, J.; Ficzere, I. Hidden contributors to uncertainty and accuracy of results of residue analysis. Accredit. Qual. Assur. 2011, 16, 3–11. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Han, L.; Sapozhnikova, Y. Use of a quality control approach to assess measurement uncertainty in the comparison of sample processing techniques in the analysis of pesticide residues in fruits and vegetables. Anal. Bioanal. Chem. 2018, 410, 5465–5479. [Google Scholar] [CrossRef]
- Lehotay, S.J.; Michlig, N.; Lightfield, A.R. Assessment of test portion sizes after sample comminution with liquid nitrogen in an improved high-throughput method for analysis of pesticide residues in fruits and vegetables. J. Agric. Food Chem. 2020, 68, 1468–1479. [Google Scholar] [CrossRef]
- Ramsey, C.A.; Wagner, C. Sample quality criteria. J. AOAC Int. 2015, 98, 265–268. [Google Scholar] [CrossRef]
- FAO. Recommended method of sampling for the determination of pesticide residues for compliance with mrls CXG33-1999. Available online: https://www.fao.org/fao-who-codexalimentarius/sh-proxy/en/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXG%2B33-1999%252FCXG_033e.pdf (accessed on 5 November 2022).
- FAO. CAC guidelines on performance criteria for methods of analysis for the determination of pesticide residues in food and feed CXG90-2017. Available online: https://www.fao.org/fao-who-codexalimentarius/sh-proxy/en/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXG%2B90-2017%252FCXG_090e.pdf (accessed on 5 November 2022).
- Bettencourt da Silva, R.J.N.; Santos, J.R.; Filomena, M.; Camoes, G.F.C. Evaluation of the analytical method performance for incurred samples. Anal. Chim. Acta 2003, 485, 241–252. [Google Scholar] [CrossRef]
- Fussel, R.J.; Hetmanski, M.T.; MacArthur, R.; Findlay, D.; Smith, F.; Ambrus, Á.; Brodesser, P.J. Measurement uncertainty associated with sample processing of oranges and tomatoes for pesticide residue analysis. J. Agric. Food Chem. 2007, 55, 1062–1070. [Google Scholar] [CrossRef]
- Suszter, K.G.; Ambrus, Á. Testing the efficiency of extraction of incurred residues from soil with optimized multi-residue method. J. Env. Sci. Health B 2017, 52, 547–556. [Google Scholar] [CrossRef]
- Han, L.; Lehotay, S.J.; Sapozhnikova, Y. Use of an efficient measurement uncertainty approach to compare room temperature and cryogenic sample processing in the analysis of chemical contaminants in foods. J. Agric. Food Chem. 2017, 66, 4986–4996. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cho, Y.-J.; Song, J.-W.; Kim, Y.-J.; Seo, J.-S.; Kim, J.-H. Residue behavior of methoxyfenozide and pymetrozine in Chinese cabbage and their health risk assessment. Foods 2022, 11, 2995. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, Á. Variability of pesticide residues in crop units. Pest Manag. Sci. 2006, 62, 693–714. [Google Scholar] [CrossRef] [PubMed]
- Horváth, Z.; Ambrus, Á.; Mészáros, L.; Braun, S. Characterization of distribution of pesticide residues in crop units. J. Environ. Sci. Health B 2013, 48, 615–625. [Google Scholar] [CrossRef]
- Ambrus, Á. Estimation of sampling uncertainty for determination of pesticide residues in plant commodities. J. Env. Sci. Health B 2009, 44, 627–639. [Google Scholar] [CrossRef]
- Farkas, Z.; Cook, J.M.; Ambrus, Á. Estimation of uncertainty of measured residues and testing compliance with MRLs. In Food Safety Assessment of Pesticide Residues, 1st ed.; Ambrus, Á., Hamilton, D., Eds.; World Scientific: Hackensack, NJ, USA, 2017; pp. 404–466. [Google Scholar]
- FAO. Portion of Commodities to Which Maximum Residue Limits Apply and Which is Analyzed CAC/GL 41-1993. Available online: www.fao.org/fao-who-codexalimentarius/sh-proxy/en/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXG%2B41-1993%252FCXG_041e.pdf (accessed on 5 November 2022).
- Omeroglu, P.Y.; Ambrus, Á.; Boyacioglu, D.; Majzik, E.S. Uncertainty of the sample size reduction step in pesticide residue analysis of large-sized crops. Food Addit. Contam. Part A 2013, 30, 116–126. [Google Scholar] [CrossRef]
- Ambrus, Á.; Szemánné-Dobrik, H.; Domák, Z.; Kerekes, K. Sources of random variation of pesticide residue analytical results. J. AOAC Int. 2021, 104, 526–538. [Google Scholar] [CrossRef]
- Gy, P. Sampling for Analytical Purposes; John Wiley & Sons Ltd.: Chichester, UK, 1999; p. 71. [Google Scholar]
- Minkkinen, P. Practical applications of sampling theory. Chemometr. Intell. Lab. Syst. 2004, 74, 85–94. [Google Scholar] [CrossRef]
- Hill, A.R.C.; Harris, C.A.; Warburton, A.G. Effect of sample processing on pesticide residues in fruit and vegetables. In Principles of Method Validation; Fajgelj, A., Ambrus, Á., Eds.; Royal Society of Chemistry: Cambridge, UK, 2000; pp. 41–48. [Google Scholar]
- Ambrus, Á.; Suszter, G. Quality Control and Quality Assurance. In Analysis of Pesticides in Food and Environmental Samples, 2nd ed.; Toledo, J., Ed.; CRC Press: Roca Baton, FL, USA, 2019; pp. 135–173. [Google Scholar]
- Ritter, L.S.; Lynn, K.J.; Wujcik, C.E.; Buchholz, L.M. Interlaboratory assessment of cryomilling sample preparation for residue analysis. J. Agric. Food Chem. 2015, 63, 4405–4408. [Google Scholar] [CrossRef]
- Ritter, L.S.; Wujcik, C.E. Novel two-stage fine milling enables high-throughput determination of glyphosate residues in raw agricultural commodities. J. AOAC Internat. 2018, 101, 867–875. [Google Scholar] [CrossRef]
- Roussev, M.; Lehotay, S.J.; Pollaehne, J. Cryogenic sample processing with liquid nitrogen for effective and efficient monitoring of pesticide residues in foods and feeds. J. Agric. Food Chem. 2019, 67, 9203–9209. [Google Scholar] [CrossRef]
- Thi Le, L.H.; Tran-Lam, T.T.; Cam, T.Q.; Nguyen, T.N.; Dao, Y.N. Pesticides in edible mushrooms in Vietnam. Food Addit. Contam. B 2021, 14, 139–148. [Google Scholar] [CrossRef]
- Ly, T.-K.; Behra, P.; Nhu-Trang, T.-T. Quantification of 397 pesticide residues in different types of commercial teas: Validation of high accuracy methods and quality assessment. Food Chem. 2022, 370, 130986. [Google Scholar] [CrossRef] [PubMed]
- FAO. Submission and evaluation of pesticide residues data for the estimation of maximum residue levels in food and feed. In FAO Plant Production and Protection paper 225; FAO: Rome, Italy, 2016; p. 298. [Google Scholar]
- FAO. Acibenzolar-S-methyl, benzovindiflupyr, flupyradifurone, penconazole, sulfoxaflor. In Pesticide Residues in Food–2016 Evaluations; FAO Plant Production and Protection Paper 231; FAO: Rome, Italy, 2017; p. 2403. [Google Scholar]
- FAO. Ethephon, flutriafol, fluxapyroxad. In Pesticide Residues in Food–2015 Evaluations; FAO Plant Production and Protection Paper 226; FAO: Rome, Italy, 2016; p. 1629. [Google Scholar]
- FAO. Pesticide Residues in Food—Reports. FAO Plant Production and Protection Papers Series. Available online: http://www.fao.org/agriculture/crops/core-themes/theme/pests/jmpr/jmpr-rep/en/ (accessed on 7 November 2022).
- European Commission. Download MRLs Data. Available online: https://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/start/screen/mrls/download (accessed on 5 November 2022).
- European Commission. Technical Guideline on the Evaluation of Extraction Efficiency of Residue Analytical Methods, SANTE/2017/10632 Rev. 4. 2022. Available online: https://food.ec.europa.eu/system/files/2022-02/pesticides_mrl_guidelines_wrkdoc_2017-10632.pdf (accessed on 10 November 2022).
- Ferrer, A.C. Personal communication 2022, EU-RT-FV17 Preliminary Results. 2022; unpublished. [Google Scholar]
- Ellison, S.L.R.; Williams, A. (Eds.) EURACHEM/CITAC Guide: Quantifying Uncertainty in Analytical Measurement, 3rd ed.; CITAC: Kowloon Bay, Hong Kong, 2012; Available online: https://www.eurachem.org/index.php/publications/guides/quam (accessed on 5 November 2022).
- Ambrus, Á.; Kamirán, Á.H.; Kötelesné-Suszter, G.; Németh, A.; Solymosné, M.E. Accuracy of analytical standard solutions and uncertainty in their nominal concentrations. J. Food Investig. 2017, 63, 1398–1421. [Google Scholar]
- ISO 5725-2:2019; Accuracy (Trueness and Precision) of Measurement Methods and Results Part 2: Basic Method for the Determination of Repeatability and Reproducibility of a Standard Measurement Method. 2019. Available online: www.iso.org/standard/69419.html (accessed on 5 November 2022).
- Ambrus, Á; Noonan, G.; Németh, A.; Kötelesné-Suszter, G.; Anastassiades, M.; Kamirán, Á.H.; Solymosné, M.E. Testing the accuracy of analytical standard solutions used for quantitative determination of pesticide residues. J. AOAC Internat. 2017, 100, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Ambrus, A.; Lantos, J.; Visi, E.; Csatlós, I.; Sárvári, L. General method for the determination of pesticide residues in samples of plant origin, soil and water, i extraction and cleanup. J. Assoc. Off. Anal. Chem. 1981, 64, 733–742. [Google Scholar] [CrossRef]
- Ambrus, Á.; Buczkó, J.; Hamow, K.Á.; Juhász, V.; Solymosné Majzik, E.; Szemánné Dobrik, H.; Szitás, R. Contribution of sample processing to variability and accuracy of the results of pesticide residue analysis in plant commodities. J. Agric. Food Chem. 2016, 64, 6071–6081. [Google Scholar] [CrossRef] [PubMed]
- Fussell, R.J.; Jackson Addie, K.; Reynolds, S.L.; Wilson, M.F. Assessment of the stability of pesticides during cryogenic sample processing. 1. Apples. J. Agric. Food Chem. 2002, 50, 441–448. [Google Scholar] [CrossRef]
- Magnusson, B.; Örnemark, U. Eurachem Guide: The Fitness for Purpose of Analytical Methods–a Laboratory Guide to Method Validation and Related Topics, 2nd ed. EURACHEM. 2014. Available online: https://www.eurachem.org/images/stories/Guides/pdf/MV_guide_2nd_ed_EN.pdf (accessed on 5 November 2022). ISBN 978-91-87461-59-0.
- Thompson, M.; Howarth, R.J. The frequency distribution of analytical error. Analyst 1980, 105, 1188–1195. [Google Scholar] [CrossRef]
- ISO/IEC 17025:2017; General Requirements for the Competence of Testing and Calibration Laboratories. International Organization for Standardization: Geneva, Switzerland, 2018.
- Youden, W.J. Statistical Techniques for Collaborative Tests; AOAC: Washington, DC, USA, 1967. [Google Scholar]
- Anderson, L.R. Practical Statistics for Analytical Chemists. Appendix D12; Van Nostrand Reinhold Company: New York, NY, USA, 1987; ISBN 0442209738. [Google Scholar]
- Anastassiades, M.; Maštovská, K.; Lehotay, S.J. Evaluation of analyte protectants to improve gas chromatographic analysis of pesticides. J. Chromatogr. A 2003, 1015, 163–184. [Google Scholar] [CrossRef]
- Maštovská, K.; Lehotay, S.J.; Anastassiades, M. Combination of analyte protectants to overcome matrix effects in routine GC analysis of pesticide residues in food matrixes. Anal. Chem. 2005, 77, 8129–8137. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Ramos, R.; Lehotay, S.J.; Michlig, N.; Socas-Rodríguez, B.; Rodríguez-Delgado, M.Á. Critical review and re-assessment of analyte protectants in gas chromatography. J. Chromatogr. A 2020, 1632, 461596. [Google Scholar] [CrossRef] [PubMed]
- Soboleva, E.; Ambrus, Á. Application of a system suitability test for quality assurance and performance optimisation of a gas chromatographic system for pesticide residue analysis. J. Chromatogr. A 2004, 1027, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Miller J., N. Statistics and Chemometrics for Analytical Chemistry, 6th ed.; Ellis Horwood PTR, Prentice Hall Pearson: Harlow, UK, 2010; p. 297. ISBN 978-0-273-73042-2. [Google Scholar]





 and
and  indicate that the acceptance criterion (Δrd ≤ 10%) can or cannot be confirmed, respectively. Taken with permission from [103].
and indicate that the acceptance criterion (Δrd ≤ 10%) can or cannot be confirmed, respectively. Taken with permission from [103].
indicate that the acceptance criterion (Δrd ≤ 10%) can or cannot be confirmed, respectively. Taken with permission from [103].
and indicate that the acceptance criterion (Δrd ≤ 10%) can or cannot be confirmed, respectively. Taken with permission from [103].







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
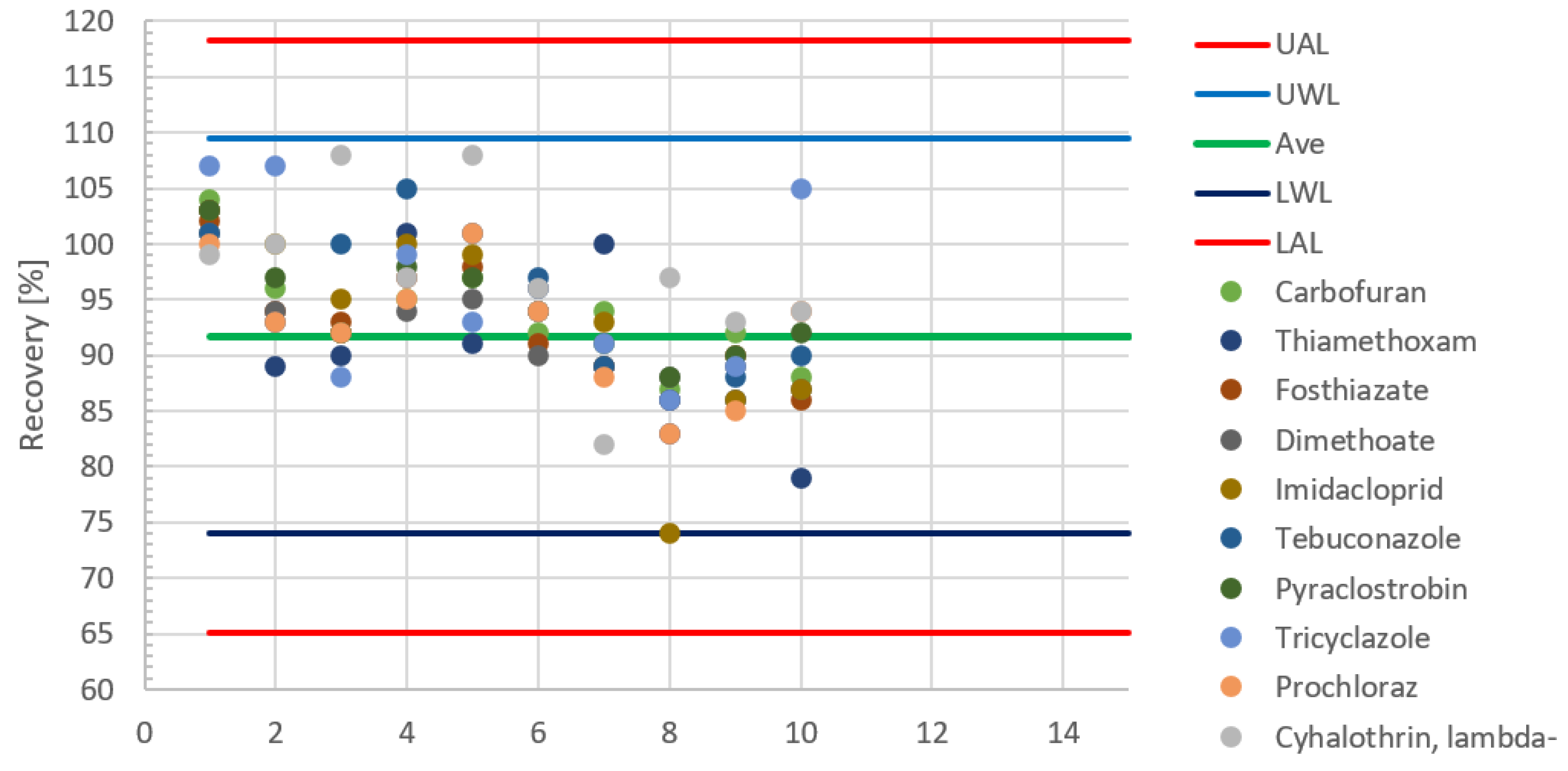{kind=link}
{kind=link}
{kind=link}
{kind=link}
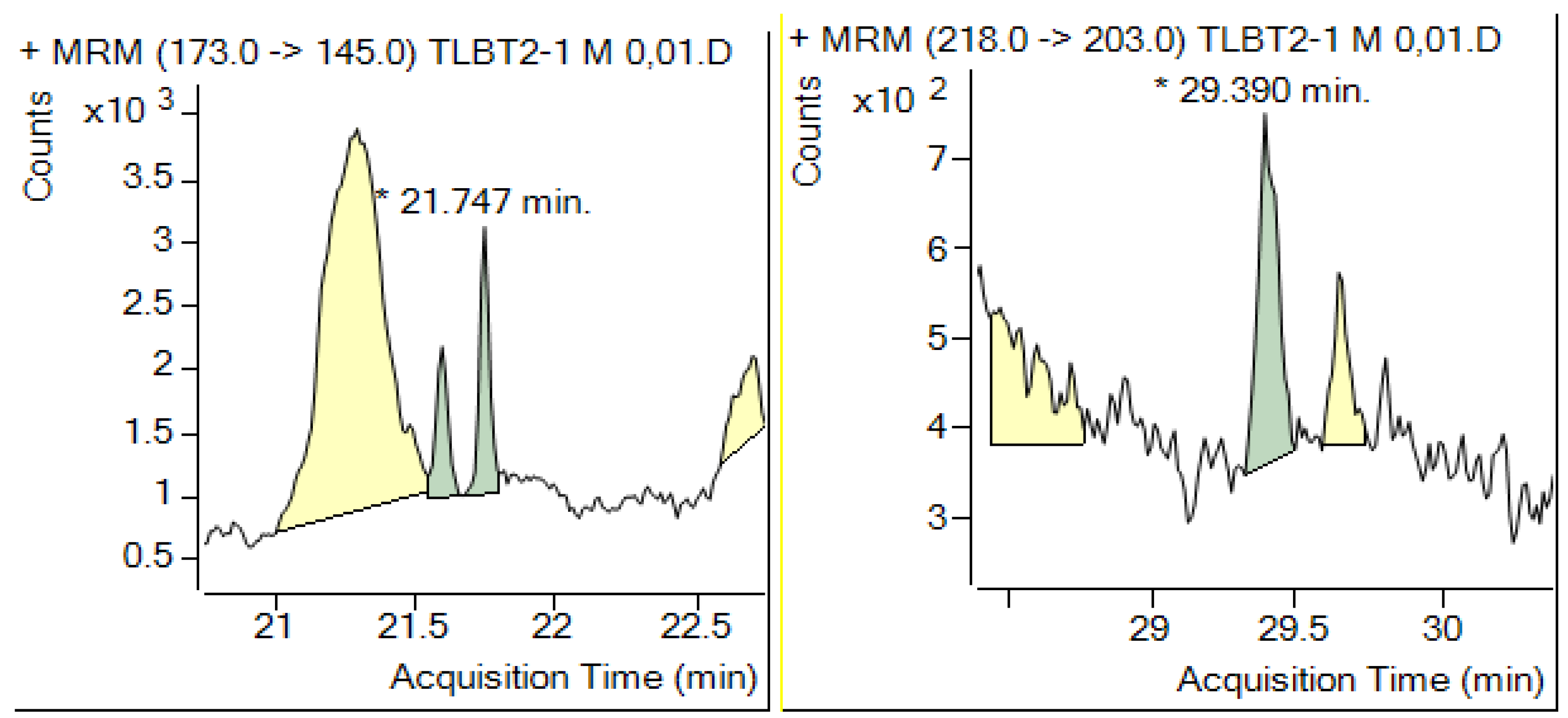{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Potential Sources of Errors | ||
|---|---|---|
| Operation | Random | Systematic |
| Sampling | Sample size; heterogeneous distribution of analyte; varying temperature during shipping and storage | Sampling target selection; sampling plan and method; degradation, evaporation of analyte; contamination of the sample; mislabeling |
| Selection of the portion of commodity to be analyzed | Inconsistent preparation of sample portion | Wrong part of the sample selected for extraction, |
| Sample size reduction, subsampling | Subsample does not represent the composition of the laboratory sample | primary samples are not proportionally represented |
| Comminution of selected sample portions | Particle size distribution in the homogenate; varying temperature and duration of comminution | Decomposition, evaporation of analytes |
| Test portion selection | Test portion does not represent the comminuted sample matrix | |
| Extraction | Varying intensity and temperature of extraction | Efficiency of extraction |
| Clean-up | Variation in the composition (e.g., water, fat, and sugar content) of sample materials; | Loss of analyte |
| Qualitative/quantitative determination of residues | Changing the retention time—shifting mass acquisition window; linearity and confidence intervals of calibration | Deviation from residue definition; missing analytes present in targeted or non-targeted analyses; high LOD; inaccurate standard solutions; matrix effect |
| CVSp | ||
|---|---|---|
| mL [g] | >5000 | 1000 |
| Tp [g] | ||
| 1 | 0.387 | 0.387 |
| 2 | 0.274 | 0.274 |
| 5 | 0.173 | 0.173 |
| 10 | 0.122 | 0.122 |
| 15 | 0.100 | 0.099 |
| 25 | 0.077 | 0.076 |
| Flupyradifurone [4-[(6-chloro-3-pyridylmethyl)(2,2-difluoroethyl)amino]furan2(5H)-one | |
| definition of the residue (for compliance with MRLs) for plant commodities | flupyradifurone |
| definition of the residue (for dietary risk assessment) for plant commodities | sum of flupyradifurone, difluoroacetic acid and 6-chloronicotinic acid, expressed as parent equivalents |
| Fluxapyroxad [3-(difluoromethyl)-1-methyl-N-(3′,4′,5′-trifluoro [1,1′-biphenyl]-2-yl)-1H-pyrazole-4-carboxamide] | |
| definition of the residue (for compliance with the MRL for plant and animal commodities) | fluxapyroxad |
| definition of the residue for estimation of dietary intake for plant commodities | sum of fluxapyroxad and 3-(difluoromethyl)-N-(3′,4′,5′-trifluoro[1,1′-biphenyl]-2-yl)-1H-pyrazole-4-carboxamide (M700F008) and 3-(difluoromethyl)- 1-(ß-D-glucopyranosyl)-N-(3′,4′,5′-triflurobipheny-2-yl)-1Hpyrzaole-4-carboxamide (M700F048) and expressed as parent equivalents |
| for estimation of dietary intake for animal commodities | sum of fluxapyroxad and 3-(difluoromethyl)-N-(3′,4′,5′-trifluoro[1,1′-biphenyl]-2-yl)-1H-pyrazole-4-carboxamide (M700F008) expressed as parent equivalents; the residue is fat soluble |
| Bupirimate | Carbendazim | Diazinon | Difenoconazole | Diflubenzuron | Methoxyfenozide | Pendimethalin | Permethrin | Spinosad | Thiabendazole | Trifloxystrobin | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Certified conc. mg/L | 5.00 | 5.00 | 5.04 | 18.99 | 18.96 | 14.95 | 4.97 | 15.05 | 15.03 | 19.04 | 19.00 |
| No. Lab | 33 | 31 | 36 | 34 | 25 | 30 | 35 | 32 | 30 | 32 | 33 |
| Accurate | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Rel dif.% Min | −74.2 | −86.6 | −41.5 | −43.7 | −40.9 | −51.5 | −59.8 | −54.1 | −36.1 | −32.5 | −36.0 |
| Rel dif.% Max b | 40.0 | 164 | 202 | 36.9 | 129 | 107 | 28.0 | 73.4 | 91.0 | 116 | 118 |
| No ≥ 10% | 17 | 23 | 18 | 19 | 16 | 18 | 19 | 23 | 19 | 16 | 19 |
| Vol. Flasks | Specification | CVT | CVRfil | CVRexp |
|---|---|---|---|---|
| 25 mL | ±0.03 mL | 4.899 × 10−4 | 7.30 × 10−3 | 7.32 × 10−3 |
| 50 mL | ±0.05 mL | 4.082 × 10−4 | 7.59 × 10−4 | 8.61 × 10−4 |
| C0 mg/mL | CVR | Effective Concentration | Deviation 1 from C0 [%] | ||||
|---|---|---|---|---|---|---|---|
| Cmin | Cave | Cmax | Min | Average | Max | ||
| 1 | 0.0079 | 0.9840 | 1.0044 | 1.0084 | −1.6% | 0.44 | 0.84 |
| 0.005 | 0.0086 | 0.0050 | 0.0052 | 0.0053 | 0% | 4.00 | 6.00 |
| 0.001 | 0.0103 | 0.0010 | 0.0011 | 0.0011 | 0% | 10.00 | 10.0 |
| Standard A1 | Standard A2 | Standard B1 | Standard B2 | |
|---|---|---|---|---|
| 121315 | 112823 | 123453 | 114811.3 | |
| 121525 | 112813 | 131282 | 122092.3 | |
| 121310 | 113000 | 123456 | 114814.1 | |
| 121401 | 113121 | 124356 | 115651.1 | |
| 121392 | 112802 | 123451 | 114809.4 | |
| Ave | 121388.6 | 112911.8 | 125199.6 | 116435.6 |
| Δrd | 7.2% | 7.3% | ||
| CV | 0.000718 | 0.001262 | 0.027337 | 0.027337 |
| CVp | 0.001027 | 0.027337 | ||
| |
and indicate that the acceptance criterion (Δrd ≤ 10%) can or cannot be confirmed, respectively.| Recovery Tests with 0.2 mg/kg Spike | Survived Residues [mg/kg] | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Residues Measured [mg/kg] | |||||||||
| Bu | Ch | Etri | Etox | Bu | Ch | Etri | Etox | ||
| 0.177 | 0.165 | 0.137 | 0.176 | 0.161 | 0.157 | 0.122 | 0.139 | ||
| 0.186 | 0.182 | 0.156 | 0.176 | 0.173 | 0.180 | 0.117 | 0.165 | ||
| 0.204 | 0.183 | 0.151 | 0.153 | 0.129 | 0.116 | 0.100 | 0.133 | ||
| 0.178 | 0.152 | 0.164 | 0.170 | 0.142 | 0.128 | 0.108 | 0.132 | ||
| 0.169 | 0.169 | 0.150 | 0.164 | 0.135 | 0.137 | 0.106 | 0.125 | ||
| 0.913 | 0.852 | 0.758 | 0.840 | 0.207 | 0.231 | 0.176 | 0.217 | ||
| CVA | 0.015 | 0.015 | 0.013 | 0.011 | CVL | 0.698 | 0.937 | 0.908 | 0.885 |
| Etridiazole with Bu | Etridiazole with Ch | Etoxazole with Bu | Etoxazole with Ch | |
|---|---|---|---|---|
| 0.523 | 0.502 | 0.662 | 0.636 | |
| 0.469 | 0.420 | 0.731 | 0.655 | |
| 0.533 | 0.555 | 0.788 | 0.819 | |
| 0.528 | 0.547 | 0.711 | 0.737 | |
| 0.542 | 0.500 | 0.709 | 0.654 | |
| φ | 0.519 | 0.505 | 0.720 | 0.700 |
| 0.512 | 0.710 |
| Parameter | Bu | Ch | Etri | Etox |
|---|---|---|---|---|
| Spiking 1 g test portion (5 replicates) | ||||
| Average recovery | 0.913 | 0.852 | 0.758 | 0.840 |
| CVA | 0.015 | 0.015 | 0.013 | 0.011 |
| Spiking 10 g test portions (5 replicates) | ||||
| Average recovery | 0.970 | 0.949 | 0.815 | 0.957 |
| CVA | 0.014 | 0.014 | 0.027 | 0.017 |
| Extracting 1 g portion from surface-treated tomato | ||||
| Average survived [mg/kg] | 0.148 | 0.143 | 0.111 | 0.139 |
| CVL | 0.123 | 0.175 | 0.080 | 0.110 |
| CVSp | 0.122 | 0.175 | ||
| Extracting 10 g portion from surface-treated tomato | ||||
| Average survived [mg/kg] | 0.146 | 0.146 | 0.135 | 0.145 |
| CVL | 0.055 | 0.098 | 0.082 | 0.125 |
| CVSp | 0.053 | 0.097 | ||
| Test No 1 | Original Test Portion | Retained Test Portion | |||||
|---|---|---|---|---|---|---|---|
| Sample code | Date of anal. 2 | Residue/commodity | Test portion Code 3 | Date of anal. | Residue [mg/kg] | ||
| Name | [mg/kg] | ||||||
| 1 | M261 | 22 August 2022 | Bupirimate/ orange | 0.205 | M261/1 | 29 August 2022 | 0.216 |
| 2 | M261/2 | 6 September 2022 | 0.210 | ||||
| 3 | M261/3 | 14 September 2022 | 0.195 | ||||
| 4 | M283 | 15 September 2022 | Lufenuron/ pepper | 0.52 | M283/1 | 22 September 2022 | 0.75 |
| 5 | M283/2 | 26 September 2022 | 0.45 | ||||
| 6 | M283/3 | 3 October 2022 | 0.50 | ||||
| 7 | M283/4 | 10 October 2022 | 0.68 | ||||
| Sdrr | R2 |
|---|---|
| 0.042 | 0.9937 |
| 0.061 | 0.9976 |
| 0.085 | 0.9988 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ambrus, Á.; Doan, V.V.N.; Szenczi-Cseh, J.; Szemánné-Dobrik, H.; Vásárhelyi, A. Quality Control of Pesticide Residue Measurements and Evaluation of Their Results. Molecules 2023, 28, 954. https://doi.org/10.3390/molecules28030954
Ambrus Á, Doan VVN, Szenczi-Cseh J, Szemánné-Dobrik H, Vásárhelyi A. Quality Control of Pesticide Residue Measurements and Evaluation of Their Results. Molecules. 2023; 28(3):954. https://doi.org/10.3390/molecules28030954
Chicago/Turabian StyleAmbrus, Árpád, Vy Vy Ngoc Doan, Júlia Szenczi-Cseh, Henriett Szemánné-Dobrik, and Adrienn Vásárhelyi. 2023. "Quality Control of Pesticide Residue Measurements and Evaluation of Their Results" Molecules 28, no. 3: 954. https://doi.org/10.3390/molecules28030954