
Glutathione-Related Enzymes and Proteins: A Review

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
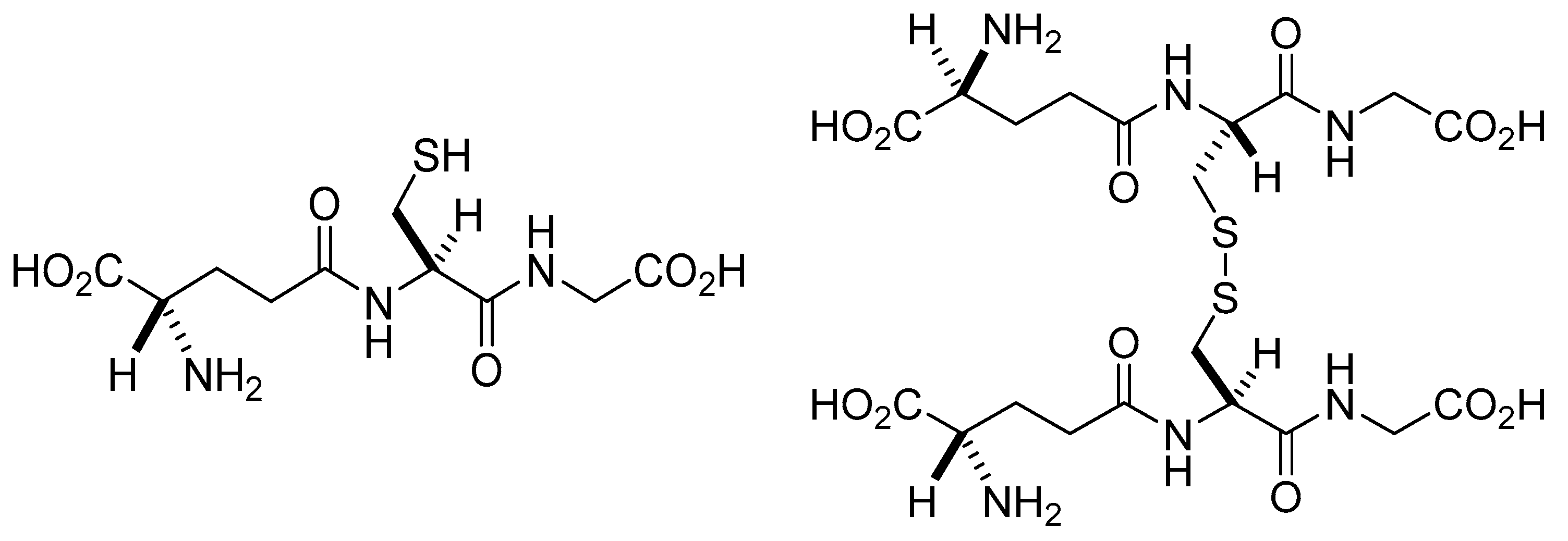
2. Glutathione
2.1. The Role of the Liver in Glutathione Synthesis and Distribution
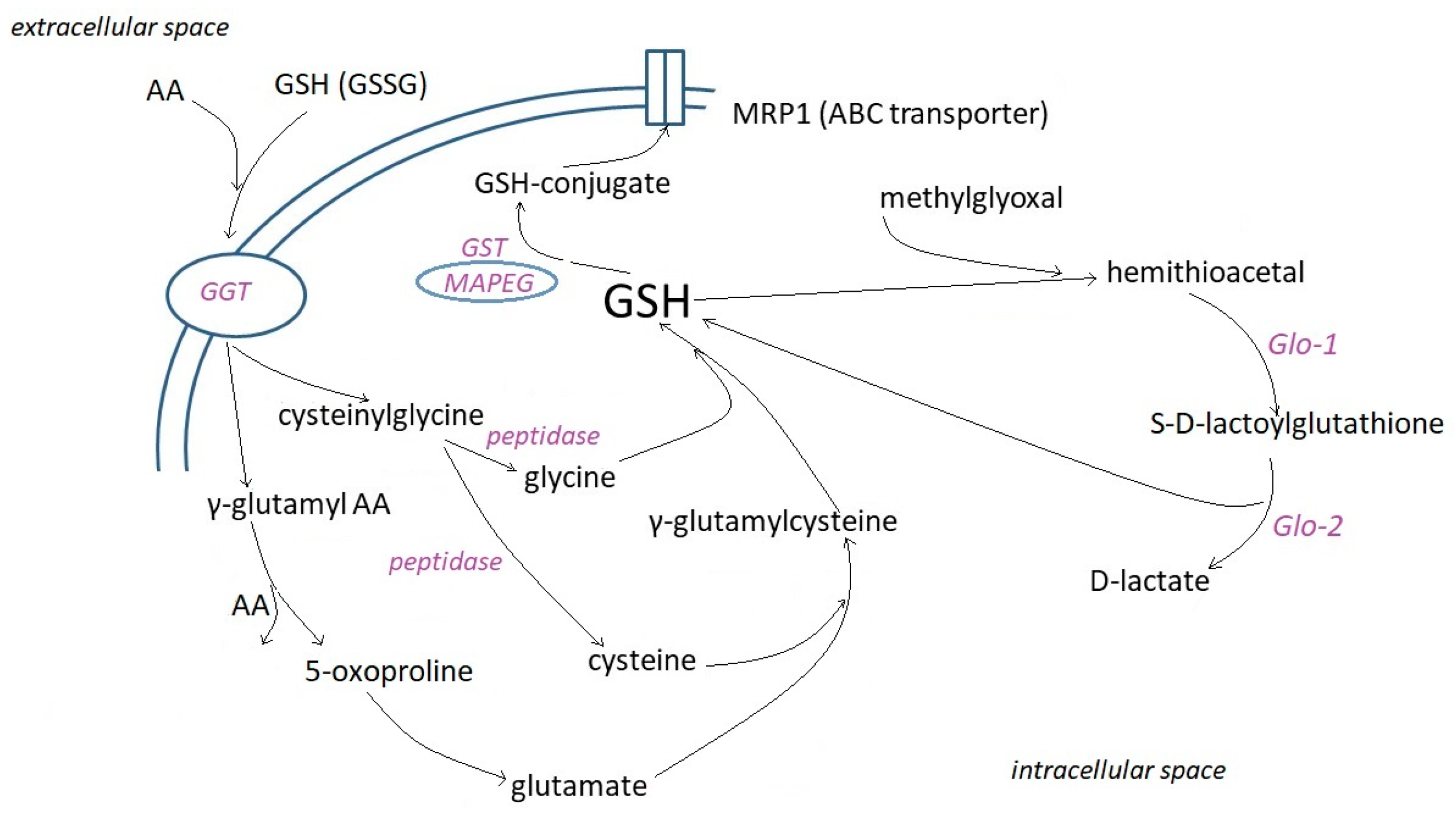
2.2. Cell Uptake and Metabolism of Glutathione
2.3. Intracellular Distribution and Functions of GSH
2.4. Acid–Base Properties
2.5. Antioxidant Properties
2.6. Redox Signaling Properties
2.7. Reactions with Electrophilic Xenobiotics
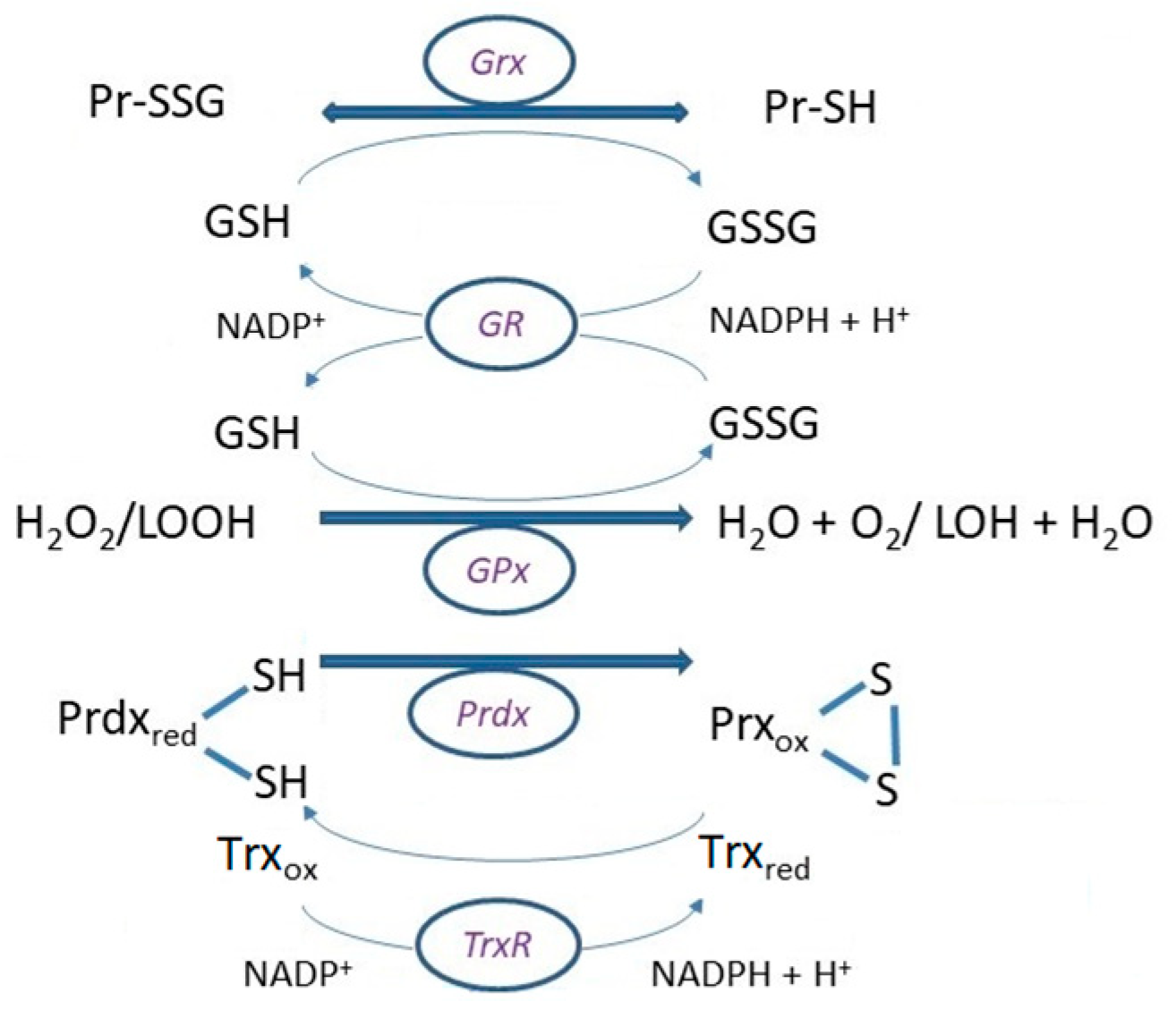
3. The Glutathione Peroxidase System
4. Glutaredoxins (Grx)
4.1. Glutathionylation
4.2. Deglutathionylation
5. Peroxiredoxins (Prdx)
6. Glutathione-S-Transferases (GST)
7. Glyoxylases (Glo)
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lillig, C.H.; Berndt, C. Preface. Cellular functions of glutathione. Biochim. Biophys. Acta 2013, 1830, 3137–3138. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, F.G. On an autoxidizable constituent of the cell. Biochem. J. 1921, 15, 286–305. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, F.G. On glutathione, a reinvestigation. J. Biol. Chem. 1929, 84, 269–320. [Google Scholar] [CrossRef]
- Hunter, G.; Eagles, B.A. Glutathione. A critical study. J. Biol. Chem. 1927, 72, 147–166. [Google Scholar] [CrossRef]
- Simoni, R.D.; Hill, R.L.; Vaughan, M. The discovery of glutathione by F. Gowland Hopkins and the beginning of biochemistry at Cambridge University. J. Biol. Chem. 2002, 277, 27–28. [Google Scholar] [CrossRef]
- Alanazi, A.M.; Mostafa, G.A.E.; Al-Badr, A.A. Glutathione. Profiles Drug Subst. Excip. Relat. Methodol. 2015, 40, 43–158. [Google Scholar]
- Lash, L.H. Mitochondrial glutathione transport: Physiological, pathological and toxicological implications. Chem. Biol. Interact. 2006, 163, 54–67. [Google Scholar] [CrossRef]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta 2013, 1830, 3143–3153. [Google Scholar] [CrossRef]
- Ballatori, N.; Krance, S.M.; Notenboom, S.; Shi, S.; Tieu, K.; Hammond, C.L. Glutathione dysregulation and the etiology and progression of human diseases. Biol. Chem. 2009, 390, 191–214. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Jozefczak, M.; Remans, T.; Vangronsveld, J.; Cuypers, A. Glutathione is a key player in metal-induced oxidative stress defenses. Int. J. Mol. Sci. 2012, 13, 3145–3175. [Google Scholar]
- Dickinson, D.A.; Forman, H.J. Cellular glutathione and thiols metabolism. Biochem. Pharmacol. 2002, 64, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002, 348, 93–112. [Google Scholar] [PubMed]
- Alberty, R.A. Standard apparent reduction potentials of biochemical half reactions and thermodynamic data on the species involved. Biophys. Chem. 2004, 111, 115–122. [Google Scholar] [CrossRef] [PubMed]
- López-Lázaro, M. A new view of carcinogenesis and an alternative approach to cancer therapy. Mol. Med. 2010, 16, 144–153. [Google Scholar]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef]
- Bachhawat, A.K.; Thakur, A.; Kaur, J.; Zulkifli, M. Glutathione transporters. Biochim. Biophys. Acta 2013, 1830, 3154–3164. [Google Scholar] [CrossRef]
- Garcia, R.A.; Stipanuk, M.H. The splanchnic organs, liver and kidney have unique roles in the metabolism of sulfur amino acids and their metabolites in rats. J. Nutr. 1992, 122, 1693–1701. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Dominy, J.E., Jr.; Lee, J.I.; Coloso, R.M. Mammalian cysteine metabolism: New insights into regulation of cysteine metabolism. J. Nutr. 2006, 136, 1652S–1659S. [Google Scholar] [CrossRef] [PubMed]
- Ookhtens, M.; Kaplowitz, N. Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine. Semin. Liver Dis. 1998, 18, 313–329. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C.; Mato, J.M. S-adenosylmethionine in liver health, injury, and cancer. Physiol. Rev. 2012, 92, 1515–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantin, A.M.; North, S.L.; Hubbard, R.C.; Crystal, R.G. Normal alveolar epithelial lining fluid contains high levels of glutathione. J. Appl. Physiol. 1987, 63, 152–157. [Google Scholar] [CrossRef] [PubMed]
- van der Vliet, A.; O’Neill, C.A.; Cross, C.E.; Koostra, J.M.; Volz, W.G.; Halliwell, B.; Louie, S. Determination of low-molecular-mass antioxidant concentrations in human respiratory tract lining fluids. Am. J. Physiol. 1999, 276, L289–L296. [Google Scholar] [CrossRef]
- Lieberman, M.W.; Wiseman, A.L.; Shi, Z.Z.; Carter, B.Z.; Barrios, R.; Ou, C.N.; Che’vez-Barrios, P.; Wang, Y.; Habib, G.M.; Goodman, J.C.; et al. Growth retardation and cysteine deficiency in gammaglutamyl transpeptidase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 7923–7926. [Google Scholar] [CrossRef]
- Kiba, N. Enzymes in physiological samples. In Encyclopedia of Analytical Science, 2nd ed.; Worsfold, P., Poole, C.F., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; pp. 536–544. [Google Scholar]
- Griffith, O.W.; Meister, A. Translocation of intracellular glutathione to membrane-bound γ-glutamyl transpeptidase as a discrete step in the γ-glutamyl cycle: Glutathionuria after inhibition of transpeptidase. Proc. Natl. Acad. Sci. USA 1979, 76, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Njalsson, R.; Norgren, S. Physiological and pathological aspects of GSH metabolism. Acta Paediatr. 2005, 94, 132–137. [Google Scholar] [CrossRef]
- Csanaky, I.; Gregus, Z. Role of glutathione in reduction of arsenate and of gamma-glutamyltranspeptidase in disposition of arsenite in rats. Toxicology 2005, 207, 91–104. [Google Scholar] [CrossRef]
- Calvio, C.; Romagnuolo, F.; Vulcano, F.; Speranza, G.; Morelli, C.F. Evidences on the role of the lid loop of γ-glutamyltransferases (GGT) in substrate selection. Enzym. Microb. Technol. 2018, 114, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Allison, D. γ-Glutamyl transpeptidase: Kinetics and mechanism. Methods Enzymol. 1985, 113, 419–437. [Google Scholar] [PubMed]
- Lam, B.K.; Austen, K.F. Leukotriene C4 synthase: A pivotal enzyme in cellular biosynthesis of the cysteinyl leukotrienes. Prostag. Other Lipid Mediat. 2002, 68–69, 511–520. [Google Scholar] [CrossRef]
- Lu, E.; Wolfreys, F.D.; Muppidi, J.R.; Xu, Y.; Cyster, J.G. S-Geranylgeranyl-L-glutathione is a ligand for human B cell-confinement receptor P2RY8. Nature 2019, 567, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Tate, S.S.; Meister, A. Interaction of γ-glutamyl transpeptidase with amino acids, dipeptides and derivatives and analogs of glutathione. J. Biol. Chem. 1974, 249, 7593–7602. [Google Scholar] [CrossRef]
- Hanigan, M.H.; Frierson, H.F., Jr.; Swanson, P.E.; De Young, B.R. Altered expression of gamma-glutamyl transpeptidase in human tumors. Hum. Pathol. 1999, 30, 300–305. [Google Scholar] [CrossRef]
- Hanigan, M.H. Gamma-glutamyl transpeptidase: Redox regulation and drug resistance. Adv. Cancer Res. 2014, 122, 103–141. [Google Scholar]
- Lash, L.H.; Jones, D.P. Transport of glutathione by renal basal-lateral membrane vesicles. Biochem. Biophys. Res. Commun. 1983, 112, 55–60. [Google Scholar] [CrossRef]
- Lash, L.H. Renal glutathione transport: Identification of carriers, physiological functions, and controversies. Biofactors 2009, 35, 500–508. [Google Scholar] [CrossRef]
- Iantomasi, T.; Favilli, F.; Marraccini, P.; Magaldi, T.; Bruni, P.; Vincenzini, M.T. Glutathione transport system in human small intestine epithelial cells. Biochim. Biophys. Acta 1997, 1330, 274–283. [Google Scholar] [CrossRef]
- Kannan, R.; Mittur, A.; Bao, Y.; Tsuruo, T.; Kaplowitz, N. GSH transport in immortalized mouse brain endothelial cells: Evidence for apical localization of a sodium-dependent GSH transporter. J. Neurochem. 1999, 73, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Putt, D.A. Renal cellular transport of exogenous glutathione: Heterogeneity at physiological and pharmacological concentrations. Biochem. Pharmacol. 1999, 58, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H. Role of glutathione transport processes in kidney function. Toxicol. Appl. Pharmacol. 2005, 204, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Galvagni, F.; Tesei, A.; Farolfi, A.; Zanoni, M.; Pignatta, S.; Milzani, A.; Marone, I.M.; Dalle-Donne, I.; Nassini, R.; et al. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures. Free Radic. Biol. Med. 2015, 89, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinsky, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef]
- Yuan, L.; Kaplowitz, N. Glutathione in liver diseases and hepatotoxicity. Mol. Asp. Med. 2009, 30, 29–41. [Google Scholar] [CrossRef]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J. Cell Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef]
- Montero, D.; Tachibana, C.; Rahr Winther, J.; Appenzeller-Herzog, C. Intracellular glutathione pools are heterogeneously concentrated. Redox Biol. 2013, 1, 508–513. [Google Scholar] [CrossRef]
- Kojer, K.; Bien, M.; Gangel, H.; Morgan, B.; Dick, T.P.; Riemer, J. Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J. 2012, 31, 3169–3182. [Google Scholar] [CrossRef]
- López-Mirabal, H.R.; Winther, J.R. Redox characteristics of the eukaryotic cytosol. Biochim. Biophys. Acta 2008, 1783, 629–640. [Google Scholar]
- Deponte, M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim. Biophys. Acta 2013, 1830, 3217–3266. [Google Scholar] [PubMed]
- Aw, T.Y. Cellular redox: A modulator of intestinal epithelial cell proliferation. News Physiol. Sci. 2003, 18, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Van ‘t Erve, T.J.; Wagner, B.A.; Ryckman, K.K.; Raife, T.J.; Buettner, G.R. The concentration of glutathione in human erythrocytes is a heritable trait. Free Radic. Biol. Med. 2013, 65, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar]
- Bellomo, G.; Palladini, G.; Vairetti, M. Intranuclear distribution, function and fate of glutathione and glutathione-S-conjugate in living rat hepatocytes studied by fluorescence microscopy. Microsc. Res. Tech. 1997, 36, 243–252. [Google Scholar] [CrossRef]
- Markovic, J.; Borrás, C.; Ortega, A.; Sastre, J.; Viña, J.; Pallardó, F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef]
- Pallardó, F.V.; Markovic, J.; García, J.L.; Viña, J. Role of nuclear glutathione as a key regulator of cell proliferation. Mol. Asp. Med. 2009, 30, 77–85. [Google Scholar] [CrossRef]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflügers Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Lushchak, V.I. Glutathione homeostasis and functions: Potential targets for medical intervention. J. Amino Acids 2012, 2012, 736837. [Google Scholar] [CrossRef]
- Zhong, Q.; Putt, D.A.; Xu, F.; Lash, L.H. Hepatic mitochondrial transport of glutathione: Studies in isolated rat liver mitochondria and H4IIE rat hepatoma cells. Arch. Biochem. Biophys. 2008, 474, 119–127. [Google Scholar] [CrossRef]
- Kamga, C.K.; Zhang, S.X.; Wang, Y. Dicarboxylate carrier-mediated glutathione transport is essential for reactive oxygen species homeostasis and normal respiration in rat brain mitochondria. Am. J. Physiol. Cell Physiol. 2010, 299, C497–C505. [Google Scholar] [CrossRef]
- Booty, L.M.; King, M.S.; Thangaratnarajah, C.; Majd, H.; James, A.M.; Kunji, E.R.S.; Murphy, M.P. The mitochondrial dicarboxylate and 2-oxoglutarate carriers do not transport glutathione. FEBS Lett. 2015, 589, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Cione, E.; Pingitore, A.; Perri, M.; Genchi, G. Influence of all-trans-retinoic acid on oxoglutarate carrier via retinoylation reaction. Biochim. Biophys. Acta 2009, 1791, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Coll, O.; Colell, A.; García-Ruiz, C.; Kaplowitz, N.; Fernández-Checa, J.C. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology 2003, 38, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and mitochondria. Front. Pharmacol. 2014, 5, 151. [Google Scholar] [PubMed]
- Yin, F.; Sancheti, H.; Cadenas, E. Mitochondrial thiols in the regulation of cell death pathways. Antioxid. Redox Signal. 2012, 17, 1714–1727. [Google Scholar] [CrossRef]
- Bánhegyi, G.; Lusini, L.; Puskás, F.; Rossi, R.; Fulceri, R.; Braun, L.; Mile, V.; di Simplicio, P.; Mandl, J.; Benedetti, A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J. Biol. Chem. 1999, 274, 12213–12216. [Google Scholar] [CrossRef] [Green Version]
- Bulleid, N.J.; Ellgaard, L. Multiple ways to make disulfides. Trends Biochem. Sci. 2011, 36, 485–492. [Google Scholar] [CrossRef]
- Ponsero, A.J.; Igbaria, A.; Darch, M.A.; Miled, S.; Outten, C.E.; Winther, J.R.; Palais, G.; D’autreaux, B.; Delaunay-Moisan, A.; Toledano, M.B. Endoplasmic Reticulum Transport of Glutathione by Sec61 Is Regulated by Ero1 and Bip. Mol. Cell 2017, 67, 962–973. [Google Scholar] [CrossRef]
- Csala, M.; Fulceri, R.; Mandl, J.; Benedetti, A.; Banhegyi, G. Ryanodine receptor channel dependent glutathione transport in the sarcoplasmic reticulum of skeletal muscle. Biochem. Biophys. Res. Commun. 2001, 287, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Riemer, J.; Zito, E.; Chin, K.-T.; Ron, D.; Spiess, M.; Ellgaard, L. Disulphide production by Ero1 alpha-PDI relay is rapid and effectively regulated. EMBO J. 2010, 29, 3318–3329. [Google Scholar] [CrossRef]
- Aoyama, K. Glutathione in the Brain. Int. J. Mol. Sci. 2021, 22, 5010. [Google Scholar] [CrossRef]
- Aoyama, K.; Nakaki, T. Impaired glutathione synthesis in neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021–21044. [Google Scholar] [PubMed]
- von Bartheld, C.S.; Bahney, J.; Herculano-Houzel, S. The search for true numbers of neurons and glial cells in the human brain: A review of 150 years of cell counting. J. Comp. Neurol. 2016, 524, 3865–3895. [Google Scholar]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Mirzahosseini, A.; Somlyay, M.; Noszál, B. The comprehensive acid–base characterization of glutathione. Chem. Phys. Lett. 2015, 622, 50–56. [Google Scholar] [CrossRef]
- Mazák, K.; Noszál, B. Advances in microspeciation of drugs and biomolecules: Species-specific concentrations, acid-base properties and related parameters. J. Pharm. Biomed. Anal. 2016, 130, 390–403. [Google Scholar] [CrossRef]
- Bjerrum, N. Dissociation constants of polybasic acids and their application to the calculation of molecular dimensions. Z. Phys. Chem. 1923, 106, 219–242. [Google Scholar] [CrossRef]
- Noszál, B. Group constant: A measure of submolecular basicity. J. Phys. Chem. 1986, 90, 4104–4110. [Google Scholar] [CrossRef]
- Fujiwara, S.; Ishizuka, H.; Fudano, S. NMR study of amino acids and their derivatives. Chem. Lett. 1974, 3, 1281–1284. [Google Scholar] [CrossRef]
- Noszál, B.; Scheller-Krattiger, V.; Martin, R.B. A unified view of carbon bound hydrogen exchange of H(2) in imidazoles and H(8) in purine nucleosides and their metal ion complexes. J. Am. Chem. Soc. 1982, 104, 1078–1081. [Google Scholar] [CrossRef]
- Noszál, B.; Rabenstein, D.L. Nitrogen-protonation microequilibria and C(2)-deprotonation microkinetics of histidine, histamine, and related compounds. J. Phys. Chem. 1991, 95, 4761–4765. [Google Scholar] [CrossRef]
- Szakács, Z.; Noszál, B. Determination of dissociation constants of folic acid, methotrexate, and other photolabile pteridines by pressure-assisted capillary electrophoresis. Electrophoresis 2006, 27, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Orgován, G.; Tihanyi, K.; Noszál, B. NMR analysis, protonation equilibria and decomposition kinetics of tolperisone. J. Pharm. Biomed. Anal. 2009, 50, 718–723. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Baska, F.; Schretner, A.; Rácz, Á.; Noszál, B. Site-specific basicities regulate molecular recognition in receptor binding: In silico docking of thyroid hormones. Eur. Biophys. J. 2013, 42, 721–730. [Google Scholar] [CrossRef]
- Pirie, N.W.; Pinhey, K.G. The titration curve of glutathione. J. Biol. Chem. 1929, 84, 321–333. [Google Scholar] [CrossRef]
- Li, N.C.; Gawron, O.; Bascuas, G. Stability of zinc complexes with glutathione and oxidized glutathione. J. Am. Chem. Soc. 1954, 76, 225–229. [Google Scholar] [CrossRef]
- Martin, R.B.; Edsall, J.T. Glutathione: Ionization in basic solutions and molecular rearrangement in strongly acid solution. Bull. Soc. Chim. Biol. 1958, 40, 1763–1771. [Google Scholar]
- Dorcák, V.; Kręzel, A. Correlation of acid–base chemistry of phytochelatin PC2 with its coordination properties towards the toxic metal ion Cd(II). Dalton Trans. 2003, 11, 2253–2259. [Google Scholar] [CrossRef]
- Mah, V.; Jalilehvand, F. Mercury(II) complex formation with glutathione in alkaline aqueous solution. J. Biol. Inorg. Chem. 2008, 13, 541–553. [Google Scholar] [CrossRef]
- Noszál, B.; Szakács, Z. Microscopic protonation equilibria of oxidized glutathione. J. Phys. Chem. B 2003, 107, 5074–5080. [Google Scholar] [CrossRef]
- Wang, X.; Li, K.; Yang, X.D.; Wang, L.L.; Shen, R.F. Complexation of Al(III) with reduced glutathione in acidic aqueous solutions. J. Inorg. Biochem. 2009, 103, 657–666. [Google Scholar] [CrossRef]
- Gough, J.D.; Lees, W.J. Effects of redox buffer properties on the folding of a disulfide-containing protein: Dependence upon pH, thiol pKa, and thiol concentration. J. Biotechnol. 2005, 115, 279–290. [Google Scholar] [CrossRef]
- Madej, E.; Wardman, P. The oxidizing power of the glutathione thiyl radical as measured by its electrode potential at physiological pH. Arch. Biochem. Biophys. 2007, 462, 94–102. [Google Scholar] [CrossRef]
- Cigala, R.M.; Crea, F.; De Stefano, C.; Lando, G.; Milea, D.; Sammartano, S. Modeling the acid–base properties of glutathione in different ionic media, with particular reference to natural waters and biological fluids. Amino Acids. 2012, 43, 629–648. [Google Scholar] [CrossRef]
- Harris, T.K.; Turner, G.J. Structural basis of perturbed pKa values of catalytic groups in enzyme active sites. IUBMB Life 2002, 53, 85–98. [Google Scholar] [CrossRef]
- Matsui, R.; Ferran, B.; Oh, A.; Croteau, D.; Shao, D.; Han, J.; Pimentel, D.R.; Bachschmid, M.M. Redox Regulation via Glutaredoxin-1 and Protein S-Glutathionylation. Antioxid. Redox Signal. 2020, 32, 677–700. [Google Scholar] [CrossRef]
- Chatgilialoglu, C.; Bowry, V.W. Why Not Trans? Inhibited Radical Isomerization Cycles and Coupling Chains of Lipids and Alkenes with Alkane-thiols. J. Org. Chem. 2018, 83, 9178–9189. [Google Scholar] [CrossRef]
- Abedinzadeh, Z.; Gardes-Albert, M.; Ferradini, C. Kinetic study of the oxidation mechanism of glutathione by hydrogen peroxide in neutral aqueous medium. Can. J. Chem. 1989, 67, 1247–1255. [Google Scholar] [CrossRef]
- Zinatullina, K.M.; Kasaikina, O.T.; Kuz’min, V.A.; Khrameeva, N.P. Interaction of Glutathione with Hydrogen Peroxide: A Kinetic Model. Kinet. Catal. 2019, 60, 266–272. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Radical Scavenging by Thiols and the Fate of Thiyl Radicals. In Oxidative Stress and Redox Regulation; Jakob, U., Reichmann, D., Eds.; Springer: Dordrecht, The Netherlands, 2013; pp. 43–58. [Google Scholar]
- Quintana-Cabrera, R.; Bolaños, J.P. Glutathione and γ-glutamylcysteine in hydrogen peroxide detoxification. Methods Enzymol. 2013, 527, 129–144. [Google Scholar]
- Kasamatsu, S.; Nishimura, A.; Morita, M.; Matsunaga, T.; Abdul Hamid, H.; Akaike, T. Redox signaling regulated by cysteine persulfide and protein polysulfidation. Molecules 2016, 21, 1712. [Google Scholar] [CrossRef]
- Sawa, T.; Ono, K.; Tsutsuki, H.; Zhang, T.; Ida, T.; Nishida, M.; Akaike, T. Reactive cysteine persulphides: Occurrence, biosynthesis, antioxidant activity, methodologies, and bacterial persulphide signalling. Adv. Microb. Physiol. 2018, 72, 1–28. [Google Scholar]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef]
- Chauvin, J.-P.R.; Griesser, M.; Pratt, D.A. Hydropersulfides: H-atom transfer agents par excellence. J. Am. Chem. Soc. 2017, 139, 6484–6493. [Google Scholar] [CrossRef]
- Libiad, M.; Motl, N.; Akey, D.L.; Sakamoto, N.; Fearon, E.R.; Smith, J.L.; Banerjee, R. Thiosulfate sulfurtransferase-like domain-containing 1 protein interacts with thioredoxin. J. Biol. Chem. 2018, 293, 2675–2686. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, V. Chemical biology of H2S signaling through persulfidation. Chem. Rev. 2018, 118, 1253–1337. [Google Scholar]
- Mueller, E.G. Trafficking in persulfides: Delivering sulfur in biosynthetic pathways. Nat. Chem. Biol. 2006, 2, 185–194. [Google Scholar] [CrossRef]
- Kruithof, P.D.; Lunev, S.; Aguilar Lozano, S.P.; de Assis Batista, F.; Al-Dahmani, Z.M.; Joles, J.A.; Dolga, A.M.; Groves, M.R.; van Goor, H. Unraveling the role of thiosulfate sulfurtransferase in metabolic diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165716. [Google Scholar] [CrossRef] [PubMed]
- Kabil, O.; Motl, N.; Strack, M.; Seravalli, J.; Metzler-Nolte, N.; Banerjee, R. Mechanism-based inhibition of human persulfide dioxygenase by γ-glutamyl-homocysteinyl-glycine. J. Biol. Chem. 2018, 293, 12429–12439. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med. 2004, 8, 201–212. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Chock, P.B. Posttranslational modification of cysteine in redox signaling and oxidative stress: Focus on s-glutathionylation. Antioxid. Redox Signal. 2012, 16, 471–475. [Google Scholar] [CrossRef] [Green Version]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys. 2008, 477, 183–195. [Google Scholar] [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef]
- Lou, M.F. Glutathione and Glutaredoxin in Redox Regulation and Cell Signaling of the Lens. Antioxidants 2022, 11, 1973. [Google Scholar] [CrossRef]
- Pajaud, J.; Kumar, S.; Rauch, C.; Morel, F.; Aninat, C. Regulation of signal transduction by glutathione transferases. Int. J. Hepatol. 2012, 2012, 137676. [Google Scholar] [CrossRef]
- Laborde, E. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 2010, 17, 1373–1380. [Google Scholar] [CrossRef]
- Singh, R.R.; Reindl, K.M. Glutathione S-transferases in cancer. Antioxidants 2021, 10, 701. [Google Scholar] [CrossRef]
- Wang, J.Q.; Yang, Y.; Cai, C.Y.; Teng, Q.X.; Cui, Q.; Lin, J.; Assaraf, Y.G.; Chen, Z.S. Multidrug resistance proteins (MRPs): Structure, function and the overcoming of cancer multidrug resistance. Drug Resist. Updat. 2021, 54, 100743. [Google Scholar] [PubMed]
- Ercolani, L.; Scirè, A.; Galeazzi, R.; Massaccesi, L.; Cianfruglia, L.; Amici, A.; Piva, F.; Urbanelli, L.; Emiliani, C.; Principato, G.; et al. A possible S-glutathionylation of specific proteins by glyoxalase II: An in vitro and in silico study. Cell Biochem. Funct. 2016, 34, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Rydström, J. Mitochondrial NADPH, transhydrogenase and disease. Biochim. Biophys. Acta. 2006, 1757, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cereser, C.; Boget, S.; Parvaz, P.; Revol, A. Thiram-induced cytotoxicity is accompanied by a rapid and drastic oxidation of reduced glutathione with consecutive lipid peroxidation and cell death. Toxicology 2001, 163, 153–162. [Google Scholar] [CrossRef]
- Zhao, Y.; Seefeldt, T.; Chen, W.; Wang, X.; Matthees, D.; Hu, Y.; Guan, X. Effects of glutathione reductase inhibition on cellular thiol redox state and related systems. Arch. Biochem. Biophys. 2009, 485, 56–62. [Google Scholar] [CrossRef]
- Arning, J.; Dringen, R.; Schmidt, M.; Thiessen, A.; Stolte, S.; Matzke, M.; Bottin-Weber, U.; Caesar-Geertz, B.; Jastorff, B.; Ranke, J. Structure-activity relationships for the impact of selected isothiazol-3-one biocides on glutathione metabolism and glutathione reductase of the human liver cell line Hep G2. Toxicology 2008, 246, 203–212. [Google Scholar] [CrossRef]
- Franco, J.L.; Posser, T.; Mattos, J.J.; Sánchez-Chardi, A.; Trevisan, R.; Oliveira, C.S.; Carvalho, P.S.; Leal, R.B.; Marques, M.R.; Bainy, A.C.; et al. Biochemical alterations in juvenile carp (Cyprinus carpio) exposed to zinc: Glutathione reductase as a target. Environ. Res. 2008, 66, 88–89. [Google Scholar] [CrossRef]
- Maiorino, M.; Ursini, F.; Bosello, V.; Toppo, S.; Tosatto, S.C.; Mauri, P.; Becker, K.; Roveri, A.; Bulato, C.; Benazzi, L.; et al. The thioredoxin specificity of Drosophila GPx: A paradigm for a peroxiredoxin-like mechanism of many glutathione peroxidases. J. Mol. Biol. 2007, 365, 1033–1046. [Google Scholar] [CrossRef]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta 2009, 1790, 1486–1500. [Google Scholar] [CrossRef]
- Flohé, L.; Toppo, S.; Cozza, G.; Ursini, F. A comparison of thiol peroxidase mechanisms. Antioxid Redox Signal. 2011, 15, 763–780. [Google Scholar] [CrossRef]
- Hill, K.E.; Burk, R.F.; Lane, J.M. Effect of selenium depletion and repletion on plasma glutathione and glutathione-dependent enzymes in the rat. J. Nutr. 1987, 117, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E.; Awad, J.A.; Morrow, J.D.; Lyons, P.R. Liver and kidney necrosis in selenium-deficient rats depleted of glutathione. Lab. Investig. 1995, 72, 723–730. [Google Scholar] [PubMed]
- Hatfield, D.L.; Gladyshev, V.N. How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 2002, 22, 3565–3576. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H(2)O(2) accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and kinetic details of catalysis of thiol-disulfide exchange by glutaredoxins and potential mechanisms of regulation. Antioxid. Redox Signal. 2009, 11, 1059–1081. [Google Scholar] [CrossRef] [PubMed]
- Hurd, T.R.; Filipovska, A.; Costa, N.J.; Dahm, C.C.; Murphy, M.P. Disulphide formation on mitochondrial protein thiols. Biochem. Soc. Trans. 2005, 33, 1390–1393. [Google Scholar] [CrossRef]
- Aslund, F.; Berndt, K.D.; Holmgren, A. Redox potentials of glutaredoxins and other thiol-disulfide oxidoreductases of the thioredoxin superfamily determined by direct protein-protein redox equilibria. J. Biol. Chem. 1997, 272, 30780–30786. [Google Scholar] [CrossRef]
- Isakov, N.; Witte, S.; Altman, A. PICOT-HD: A highly conserved protein domain that is often associated with thioredoxin and glutaredoxin modules. Trends Biochem. Sci. 2000, 25, 537–539. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar]
- Di Simplicio, P.; Cacace, M.G.; Lusini, L.; Giannerini, F.; Giustarini, D.; Rossi, R. Role of protein -SH groups in redox homeostasis--the erythrocyte as a model system. Arch. Biochem. Biophys. 1998, 355, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Thomas, J.A. S-glutathiolated hepatocyte proteins and insulin disulfides as substrates for reduction by glutaredoxin, thioredoxin, protein disulfide isomerase, and glutathione. Arch. Biochem. Biophys. 1996, 335, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.L.; Pinto, J.T.; Callery, P.S. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin. Drug Metab. Toxicol. 2011, 7, 891–910. [Google Scholar] [CrossRef] [PubMed]
- Findlay, V.J.; Townsend, D.M.; Morris, T.E.; Fraser, J.P.; He, L.; Tew, K.D. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006, 66, 6800–6806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef]
- Fratelli, M.; Demol, H.; Puype, M.; Casagrande, S.; Eberini, I.; Salmona, M.; Bonetto, V.; Mengozzi, M.; Duffieux, F.; Miclet, E.; et al. Identification by redox proteomics of glutathionylated proteins in oxidatively stressed human T lymphocytes. Proc. Natl. Acad. Sci. USA 2002, 99, 3505–3510. [Google Scholar] [CrossRef]
- Rouhier, N.; Lemaire, S.D.; Jacquot, J.P. The role of glutathione in photosynthetic organisms: Emerging functions for glutaredoxins and glutathionylation. Annu. Rev. Plant Biol. 2008, 59, 143–166. [Google Scholar] [CrossRef]
- Nordstrand, K.; Sandström, A.; Aslund, F.; Holmgren, A.; Otting, G.; Berndt, K.D. NMR structure of oxidized glutaredoxin 3 from Escherichia coli. J. Mol. Biol. 2000, 303, 423–432. [Google Scholar] [CrossRef]
- Begas, P.; Liedgens, L.; Moseler, A.; Meyer, A.J.; Deponte, M. Glutaredoxin catalysis requires two distinct glutathione interaction sites. Nat. Commun. 2017, 8, 14835. [Google Scholar] [CrossRef]
- Ukuwela, A.A.; Bush, A.I.; Wedd, A.G.; Xiao, Z. Reduction potentials of protein disulfides and catalysis of glutathionylation and deglutathionylation by glutaredoxin enzymes. Biochem. J. 2017, 474, 3799–3815. [Google Scholar] [CrossRef]
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox Biol. 2021, 43, 101975. [Google Scholar] [PubMed]
- Lillig, C.H.; Berndt, C.; Vergnolle, O.; Lönn, M.E.; Hudemann, C.; Bill, E.; Holmgren, A. Characterization of human glutaredoxin 2 as iron-sulfur protein: A possible role as red ox sensor. Proc. Natl. Acad. Sci. USA 2005, 102, 8168–8173. [Google Scholar] [CrossRef]
- Prigge, J.R.; Coppo, L.; Martin, S.S.; Ogata, F.; Miller, C.G.; Bruschwein, M.D.; Orlicky, D.J.; Shearn, C.T.; Kundert, J.A.; Lytchier, J.; et al. Hepatocyte Hyperproliferation upon Liver-Specific Co-disruption of Thioredoxin-1, Thioredoxin Reductase-1, and Glutathione Reductase. Cell Rep. 2017, 19, 2771–2781. [Google Scholar] [CrossRef] [PubMed]
- Tamarit, J.; Bellí, G.; Cabiscol, E.; Herrero, E.; Ros, J. Biochemical characterization of yeast mitochondrial Grx5 monothiol glutaredoxin. J. Biol. Chem. 2003, 278, 25745–25751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, S.; Villalba, M.; Bi, K.; Liu, Y.; Isakov, N.; Altman, A. Inhibition of the c-Jun N-terminal kinase/AP-1 and NF-κB pathways by PICOT, a novel protein kinase C-interacting protein with a thioredoxin homology domain. J. Biol. Chem. 2000, 275, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Haunhorst, P.; Hanschmann, E.M.; Bräutigam, L.; Stehling, O.; Hoffmann, B.; Mühlenhoff, U.; Lill, R.; Berndt, C.; Lillig, C.H. Crucial function of vertebrate glutaredoxin 3 (PICOT) in iron homeostasis and hemoglobin maturation. Mol. Biol. Cell 2013, 24, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.; Pasvolsky, R.; Babichev, Y.; Braiman, A.; Witte, S.; Altman, A.; Isakov, N. PICOT binding to the polycomb group protein, EED, alters H3K27 methylation at the MYT1 PRC2 target gene. Biochem. Biophys. Res. Commun. 2019, 509, 469–475. [Google Scholar] [CrossRef]
- Pandya, P.; Isakov, N. PICOT promotes T lymphocyte proliferation by down-regulating cyclin D2 expression. World J. Immunol. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Brancaccio, D.; Gallo, A.; Mikolajczyk, M.; Zovo, K.; Palumaa, P.; Novellino, E.; Piccioli, M.; Ciofi-Baffoni, S.; Banci, L. Formation of [4Fe-4S] clusters in the mitochondrial iron-sulfur cluster assembly machinery. J. Am. Chem. Soc. 2014, 136, 16240–16250. [Google Scholar] [CrossRef]
- Banci, L.; Brancaccio, D.; Ciofi-Baffoni, S.; Del Conte, R.; Gadepalli, R.; Mikolajczyk, M.; Neri, S.; Piccioli, M.; Winkelmann, J. [2Fe-2S] cluster transfer in iron-sulfur protein biogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 6203–6208. [Google Scholar] [CrossRef]
- Nasta, V.; Giachetti, A.; Ciofi-Baffoni, S.; Banci, L. Structural insights into the molecular function of human [2Fe-2S] BOLA1-GRX5 and [2Fe-2S] BOLA3-GRX5 complexes. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2119–2131. [Google Scholar] [CrossRef] [PubMed]
- Rozell, B.; Barcena, J.A.; Martinez-Galisteo, E.; Padilla, C.A.; Holmgren, A. Immunochemical characterization and tissue distribution of glutaredoxin (thioltransferase) from calf. Eur. J. Cell Biol. 1993, 62, 314–323. [Google Scholar]
- Pai, H.V.; Starke, D.W.; Lesnefsky, E.J.; Hoppel, C.L.; Mieyal, J.J. What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid. Redox Signal. 2007, 9, 2027–2033. [Google Scholar] [CrossRef] [PubMed]
- Ukuwela, A.A.; Bush, A.I.; Wedd, A.G.; Xiao, Z. Glutaredoxins employ parallel monothiol-dithiol mechanisms to catalyze thiol-disulfide exchanges with protein disulfides. Chem. Sci. 2017, 9, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- González, R.; López-Grueso, M.J.; Muntané, J.; Bárcena, J.A.; Padilla, C.A. Redox regulation of metabolic and signaling pathways by thioredoxin and glutaredoxin in NOS-3 overexpressing hepatoblastoma cells. Redox Biol. 2015, 6, 122–134. [Google Scholar] [CrossRef] [Green Version]
- Inadomi, C.; Murata, H.; Ihara, Y.; Goto, S.; Urata, Y.; Yodoi, J.; Kondo, T.; Sumikawa, K. Overexpression of glutaredoxin protects cardiomyocytes against nitric oxide-induced apoptosis with suppressing the S-nitrosylation of proteins and nuclear translocation of GAPDH. Biochem. Biophys. Res. Commun. 2012, 425, 656–661. [Google Scholar] [CrossRef]
- Lundberg, M.; Fernandes, A.P.; Kumar, S.; Holmgren, A. Cellular and plasma levels of human glutaredoxin 1 and 2 detected by sensitive ELISA systems. Biochem. Biophys. Res. Commun. 2004, 319, 801–809. [Google Scholar] [CrossRef]
- Lundberg, M.; Johansson, C.; Chandra, J.; Enoksson, M.; Jacobsson, G.; Ljung, J.; Johansson, M.; Holmgren, A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J. Biol. Chem. 2001, 276, 26269–26275. [Google Scholar] [CrossRef]
- Johansson, C.; Lillig, C.H.; Holmgren, A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 2004, 279, 7537–7543. [Google Scholar] [CrossRef]
- Hashemy, S.I.; Johansson, C.; Berndt, C.; Lillig, C.H.; Holmgren, A. Oxidation and S-nitrosylation of cysteines in human cytosolic and mitochondrial glutaredoxins: Effects on structure and activity. J. Biol. Chem. 2007, 282, 14428–14436. [Google Scholar] [CrossRef]
- Wu, H.; Xing, K.; Lou, M.F. Glutaredoxin 2 prevents H2O2-induced cell apoptosis by protecting complex I activity in the mitochondria. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Fiorenzo, P.; Nencini, M.; Cozzolino, M.; Pesaresi, M.G.; Valle, C.; Sepe, S.; Moreno, S.; Carrì, M.T. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 2010, 19, 4529–4542. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, G.; Masip, L. Biochemistry. An overoxidation journey with a return ticket. Science 2003, 300, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Bryk, R.; Griffin, P.; Nathan, C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature 2000, 407, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Nadeem, M.S.; Khan, J.A.; Murtaza, B.N.; Muhammad, K.; Rauf, A. Purification and properties of liver catalase from water buffalo (Bubalus bubalis). South Asian J. Life Sci. 2015, 3, 51–55. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A. Multiple functions of 2-Cys peroxiredoxins, I and II, and their regulations via posttranslational modifications. Free Radic. Biol. Med. 2020, 152, 107–115. [Google Scholar] [CrossRef]
- Randall, L.; Manta, B.; Nelson, K.J.; Santos, J.; Poole, L.B.; Denicola, A. Structural changes upon peroxynitrite-mediated nitration of peroxiredoxin 2; nitrated Prx2 resembles its disulfide-oxidized form. Arch. Biochem. Biophys. 2016, 590, 101–108. [Google Scholar] [CrossRef]
- Nelson, K.J.; Perkins, A.; Van Swearingen, A.E.D.; Hartman, S.; Brereton, A.E.; Parsonage, D.; Salsbury, F.R., Jr.; Karplus, P.A.; Poole, L.B. Experimentally dissecting the origins of peroxiredoxin catalysis. Antioxid. Redox Signal. 2018, 28, 521–536. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, J.; Koruza, K.; Luo, T.; Malo Pueyo, J.; Nghia Vo, T.; Ezeriņa, D.; Messens, J. Peroxiredoxins wear many hats: Factors that fashion their peroxide sensing personalities. Redox Biol. 2021, 42, 101959. [Google Scholar]
- Monteiro, G.; Horta, B.B.; Pimenta, D.C.; Augusto, O.; Netto, L.E. Reduction of 1-Cys peroxiredoxins by ascorbate changes the thiol-specific antioxidant paradigm, revealing another function of vitamin C. Proc. Natl. Acad. Sci. USA 2007, 104, 4886–4891. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.J.; Littlejohn, J.; Yewdall, N.A.; Zhu, T.; Valéry, C.; Pearce, F.G.; Mitra, A.K.; Radjainia, M.; Gerrard, J.A. Peroxiredoxin is a Versatile Self-Assembling Tecton for Protein Nanotechnology. Biomacromolecules 2014, 15, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jang, H.H. Role of Cytosolic 2-Cys Prx1 and Prx2 in Redox Signaling. Antioxidants 2019, 8, 169. [Google Scholar] [CrossRef]
- Phalen, T.J.; Weirather, K.; Deming, P.B.; Anathy, V.; Howe, A.K.; van der Vliet, A.; Jonsson, T.J.; Poole, L.B.; Heintz, N.H. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J. Cell Biol. 2006, 175, 779–789. [Google Scholar] [CrossRef]
- Saccoccia, F.; Di Micco, P.; Boumis, G.; Brunori, M.; Koutris, I.; Miele, A.E.; Morea, V.; Sriratana, P.; Williams, D.L.; Bellelli, A.; et al. Moonlighting by different stressors: Crystal structure of the chaperone species of a 2-Cys peroxiredoxin. Structure 2012, 20, 429–439. [Google Scholar] [CrossRef]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef]
- Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Activation of the antioxidant enzyme 1-CYS peroxiredoxin requires glutathionylation mediated by heterodimerization with pi GST. Proc. Natl. Acad. Sci. USA 2004, 101, 3780–3785. [Google Scholar] [CrossRef]
- Schremmer, B.; Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Peroxiredoxins in the lung with emphasis on peroxiredoxin VI. Subcell. Biochem. 2007, 44, 317–344. [Google Scholar]
- Poole, L.B.; Nelson, K.J. Distribution and features of the six classes of peroxiredoxins. Mol. Cells 2016, 39, 53–59. [Google Scholar] [PubMed]
- Aniya, Y.; Imaizumi, N. Mitochondrial glutathione transferases involving a new function for membrane permeability transition pore regulation. Drug Metab. Rev. 2011, 43, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Kural, C.; Kocdogan, A.K.; Şimşek, G.G.; Oguztuzun, S.; Kaygın, P.; Yılmaz, I.; Bayram, T.; Izci, Y. Glutathione S-transferases and cytochrome P450 enzyme expression in patients with intracranial tumors: Preliminary report of 55 patients. Med. Princ. Pract. 2018, 28, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Raza, H. Dual localization of glutathione S-transferase in the cytosol and mitochondria: Implications in oxidative stress, toxicity and disease. FEBS J. 2011, 278, 4243–4251. [Google Scholar] [CrossRef]
- Hayes, J.D.; Strange, R.C. Glutathione S-transferase polymorphisms and their biological consequences. Pharmacology 2000, 61, 154–166. [Google Scholar] [CrossRef]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar] [CrossRef]
- Chatterjee, A.; Gupta, S. The multifaceted role of glutathione S-transferases in cancer. Cancer Lett. 2018, 433, 33–42. [Google Scholar] [CrossRef]
- Townsend, D.; Tew, K. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Pacifici, G.; Franchi, M.; Bencini, C.; Repetti, F.; Di Lascio, N.; Muraro, G. Tissue distribution of drug-metabolizing enzymes in humans. Xenobiotica 1988, 18, 849–856. [Google Scholar] [CrossRef]
- Oakley, A. Glutathione transferases: A structural perspective. Drug Metab. Rev. 2011, 43, 138–151. [Google Scholar] [CrossRef]
- Ladner, J.E.; Parsons, J.F.; Rife, C.L.; Gilliland, G.L.; Armstrong, R.N. Parallel evolutionary pathways for glutathione transferases: Structure and mechanism of the mitochon-drial class kappa enzyme rGSTK1-1. Biochemistry 2004, 43, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xia, Z.; Ding, J. Thioredoxin-like domain of human kappa class glutathione transferase reveals sequence homology and structure similarity to the theta class enzyme. Protein Sci. 2005, 14, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Nathaniel, C.; Wallace, L.A.; Burke, J.; Dirr, H.W. The role of an evolutionarily conserved cis-proline in the thioredoxin-like domain of human class Alpha glutathione transferase A1-1. Biochem. J. 2003, 372, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, H.J.; Babbitt, P.C. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry 2009, 48, 11108–11116. [Google Scholar] [CrossRef]
- Stenberg, G.; Board, P.G.; Mannervik, B. Mutation of an evolutionarily conserved tyrosine residue in the active-site of a human class alpha-glutathione transferase. FEBS Lett. 1991, 293, 153–155. [Google Scholar] [CrossRef]
- Board, P.G.; Coggan, M.; Chelvanayagam, G.; Easteal, S.; Jermiin, L.S.; Schulte, G.K.; Danley, D.E.; Hoth, L.R.; Griffor, M.C.; Kamath, A.V.; et al. Identification, characterization, and crystal structure of the omega class glutathione transferases. J. Biol. Chem. 2000, 275, 24798–24806. [Google Scholar] [CrossRef]
- Mannervik, B.; Board, P.G.; Hayes, J.D.; Listowsky, I.; Pearson, W.R. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 2005, 401, 1–8. [Google Scholar]
- Hayes, J.D.; Pulford, D.J. The glutathione S-transferase gene family: Regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–600. [Google Scholar]
- Fernández-Cañón, J.M.; Peñalva, M.A. Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J. Biol. Chem. 1998, 273, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sharma, R.; Zimniak, P.; Awasthi, Y.C. Role of α class glutathione S-transferases as antioxidant enzymes in rodent tissues. Toxicol. Appl. Pharmacol. 2002, 182, 105–115. [Google Scholar] [CrossRef]
- Yang, J.Y.; Sharma, R.; Sharma, A.; Awasthi, S.; Awasthi, Y.C. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim. Pol. 2003, 50, 319–336. [Google Scholar] [CrossRef]
- Johansson, A.S.; Mannervik, B. Human glutathione transferase A3-3, a highly efficient catalyst of double-bond isomerization in the biosynthetic pathway of steroid hormones. J. Biol. Chem. 2001, 276, 33061–33065. [Google Scholar] [CrossRef]
- Flanagan, J.U.; Smythe, M.L. Sigma-class glutathione transferases. Drug Metab. Rev. 2011, 43, 194–214. [Google Scholar] [CrossRef]
- Kanaoka, Y.; Ago, H.; Inagaki, E.; Nanayama, T.; Miyano, M.; Kikuno, R.; Fujii, Y.; Eguchi, N.; Toh, H.; Urade, Y.; et al. Cloning and crystal structure of hematopoietic prostaglandin D synthase. Cell 1997, 90, 1085–1095. [Google Scholar] [CrossRef]
- Welsch, D.J.; Creely, D.P.; Hauser, S.D.; Mathis, K.J.; Krivi, G.G.; Isakson, P.C. Molecular cloning and expression of human leukotriene-C4 synthase. Proc. Natl. Acad. Sci. USA 1994, 91, 9745–9749. [Google Scholar] [CrossRef]
- Campbell, E.; Takahashi, Y.; Abramovitz, M.; Peretz, M.; Listowsky, I. A distinct human testis and brain mu-class glutathione S-transferase. Molecular cloning and characterization of a form present even in individuals lacking hepatic type mu isoenzymes. J Biol Chem. 1990, 265, 9188–9193. [Google Scholar] [CrossRef] [PubMed]
- Dulhunty, A.; Gage, P.; Curtis, S.; Chelvanayagam, G.; Board, P. The glutathione structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J. Biol. Chem. 2001, 276, 3319–3323. [Google Scholar] [CrossRef]
- Harrop, S.J.; DeMaere, M.Z.; Fairlie, W.D.; Reztsova, T.; Valenzuela, S.M.; Mazzanti, M.; Tonini, R.; Qiu, M.R.; Jankova, L.; Warton, K.; et al. Crystal structure of a soluble form of the intracellular chloride ion channel CLIC1 (NCC27) at 1.4-A resolution. J. Biol Chem. 2001, 276, 44993–45000. [Google Scholar] [CrossRef] [PubMed]
- Wilce, M.C.; Parker, M.W. Structure and function of glutathione S-transferases. Biochim. Biophys. Acta 1994, 1205, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Menon, D.; Board, P.G. A role for glutathione transferase Omega 1 (GSTO1-1) in the glutathionylation cycle. J. Biol. Chem. 2013, 288, 25769–25779. [Google Scholar] [CrossRef]
- Board, P.G.; Anders, M.W. Glutathione transferase omega 1 catalyzes the reduction of S-(phenacyl)glutathiones to acetophenones. Chem. Res. Toxicol. 2007, 20, 149–154. [Google Scholar] [CrossRef]
- Schmuck, E.M.; Bard, P.G.; Whibread, A.K.; Tetlow, N.; Cavanaugh, J.A.; Blackburn, A.C.; Masoumi, A. Characterization of the monomethylarsonate reductase and dehydroascorbate reductase activities of omega class glutathione transferase variants. Implications for arsenic metabolism and the age-at-onset of Alzheimer’s and Parkinson’s diseases. Pharmacogenet. Genom. 2005, 15, 493–501. [Google Scholar] [CrossRef]
- Dong, S.; Sha, H.; Xu, X.; Hu, T.; Lou, R.; Li, H.; Wu, J.; Dan, C.; Feng, J. Glutathione S-transferase π: A potential role in antitumor therapy. Drug Des. Dev. Ther. 2018, 12, 3535–3547. [Google Scholar] [CrossRef]
- Townsend, D.M.; Findlay, V.L.; Tew, K.D. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005, 401, 287–307. [Google Scholar]
- Antognelli, C.; Ferri, I.; Bellezza, G.; Siccu, P.; Love, H.D.; Talesa, V.N.; Sidoni, A. Glyoxalase 2 drives tumorigenesis in human prostate cells in a mechanism involving androgen receptor and p53-p21 axis. Mol. Carcinog. 2017, 56, 2112–2126. [Google Scholar] [CrossRef]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.; Ponces Freire, A.; Cordeiro, C. The glyoxalase pathway: The first hundred years… and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef]
- Ellis, K.J. Human body composition: In vivo methods. Physiol. Rev. 2000, 80, 649–680. [Google Scholar]
- Thornalley, P.J.; Waris, S.; Fleming, T.; Santarius, T.; Larkin, S.J.; Winklhofer-Roob, B.M.; Stratton, M.R.; Rabbani, N. Imidazopurinones are markers of physiological genomic damage linked to DNA instability and glyoxalase 1-associated tumour multidrug resistance. Nucleic Acids Res. 2010, 38, 5432–5442. [Google Scholar] [CrossRef]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef]
- Sarker, M.K.; Lee, J.H.; Lee, D.H.; Chun, K.H.; Jun, H.S. Attenuation of diabetic kidney injury in DPP4-deficient rats; role of GLP-1 on the suppression of AGE formation by inducing glyoxalase 1. Aging 2020, 12, 593–610. [Google Scholar] [PubMed]
- Thornalley, P.J. The glyoxalase system in health and disease. Mol. Asp. Med. 1993, 14, 287–371. [Google Scholar]
- Birkenmeier, G.; Stegemann, C.; Hoffmann, R.; Gunther, R.; Huse, K.; Birkemeyer, C. Posttranslational modification of human glyoxalase 1 indicates redox-dependent regulation. PLoS ONE 2010, 5, e10399. [Google Scholar]
- Limphong, P.; Nimako, G.; Thomas, P.W.; Fast, W.; Makaroff, C.A.; Crowde, M.W. Arabidopsis thaliana mitochondrial glyoxalase 2–1 exhibits beta-lactamase activity. Biochemistry 2009, 48, 8491–8493. [Google Scholar] [PubMed]
- Wendler, A.; Irsch, T.; Rabbani, N.; Thornalley, P.J.; Krauth-Siegel, R.L. Glyoxalase II does not support methylglyoxal detoxification but serves as a general trypanothione thioesterase in African trypanosomes. Mol. Biochem. Parasitol. 2009, 163, 19–27. [Google Scholar] [PubMed]
- Hara, T.; Toyoshima, M.; Hisano, Y.; Balan, S.; Iwayama, Y.; Aono, H.; Fatamura, Y.; Osada, H.; Owada, Y.; Yoshikawa, T. Glyoxalase I disruption and external carbonyl stress impair mitochondrial function in human induced pluripotent stem cells and derived neurons. Transl. Psychiatry 2021, 11, 275. [Google Scholar]
- Xue, M.; Shafie, A.; Qaiser, T.; Rajpoot, N.M.; Kaltsas, G.; James, S.; Gopalakrishnan, K.; Fisk, A.; Dimitriadis, G.K.; Grammatopoulos, D.K.; et al. Glyoxalase 1 copy number variation in patients with well differentiated gastro-entero-pancreatic neuroendocrine tumours (GEP-NET). Oncotarget 2017, 8, 76961–76973. [Google Scholar] [PubMed] [Green Version]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vašková, J.; Kočan, L.; Vaško, L.; Perjési, P. Glutathione-Related Enzymes and Proteins: A Review. Molecules 2023, 28, 1447. https://doi.org/10.3390/molecules28031447
Vašková J, Kočan L, Vaško L, Perjési P. Glutathione-Related Enzymes and Proteins: A Review. Molecules. 2023; 28(3):1447. https://doi.org/10.3390/molecules28031447
Chicago/Turabian StyleVašková, Janka, Ladislav Kočan, Ladislav Vaško, and Pál Perjési. 2023. "Glutathione-Related Enzymes and Proteins: A Review" Molecules 28, no. 3: 1447. https://doi.org/10.3390/molecules28031447