
The Multifaceted MEP Pathway: Towards New Therapeutic Perspectives
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
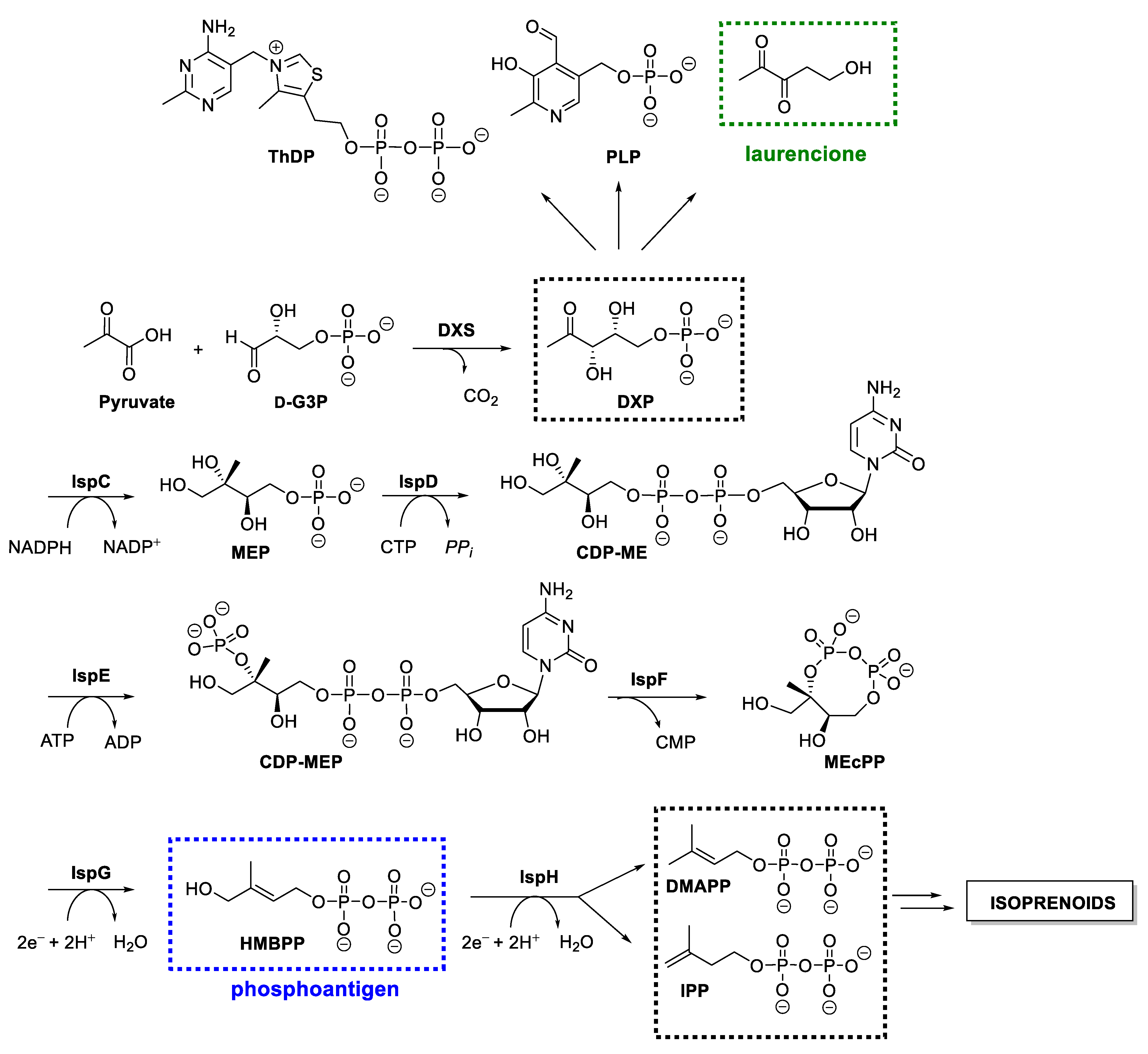
:1. Introduction
2. Antibacterials
2.1. Inhibitors of DXS
2.2. Inhibitors of DXR/IspC
2.3. Inhibitors of IspD/YgbP
2.4. Inhibitors of IspE
2.5. Inhibitors of IspF
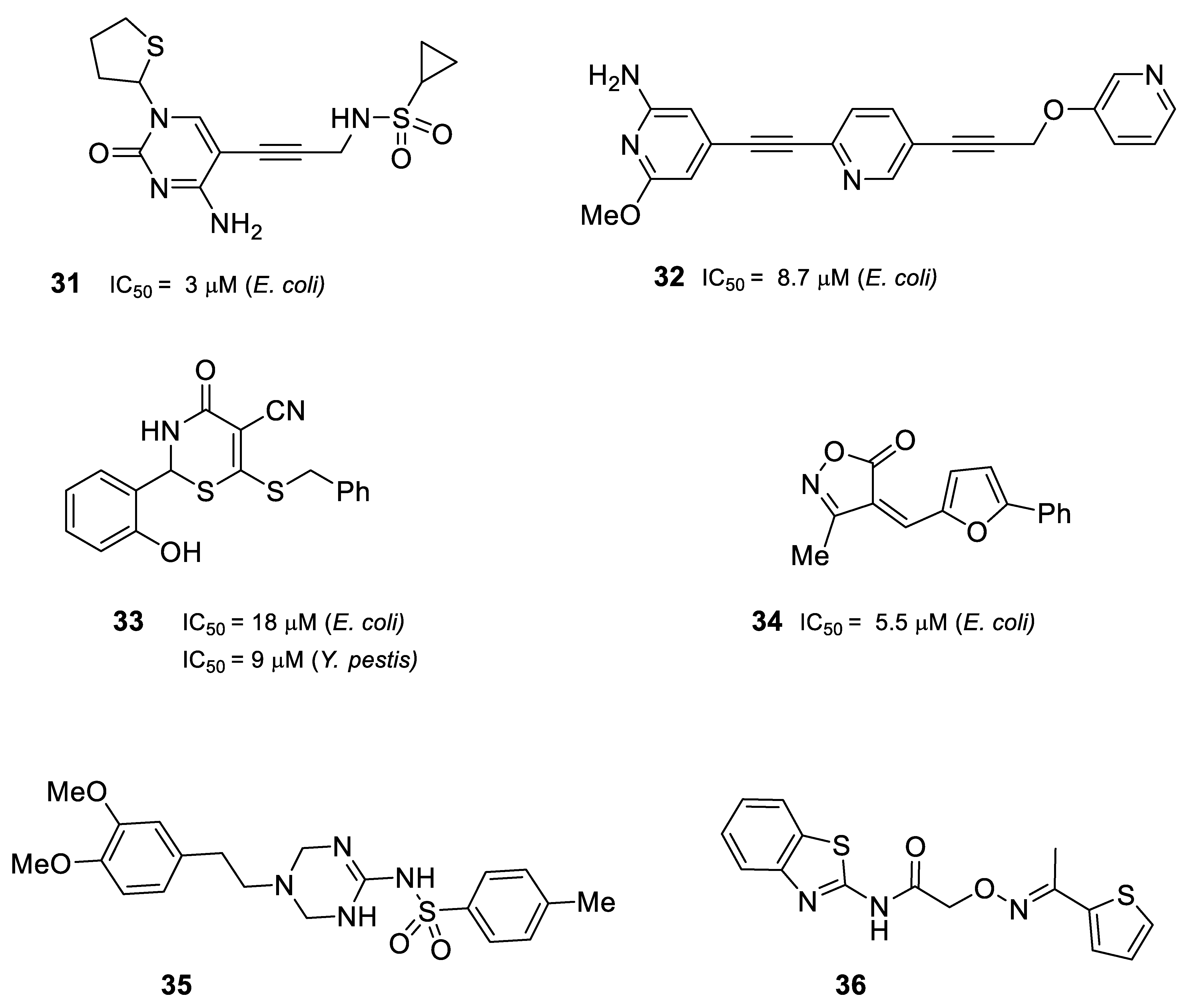
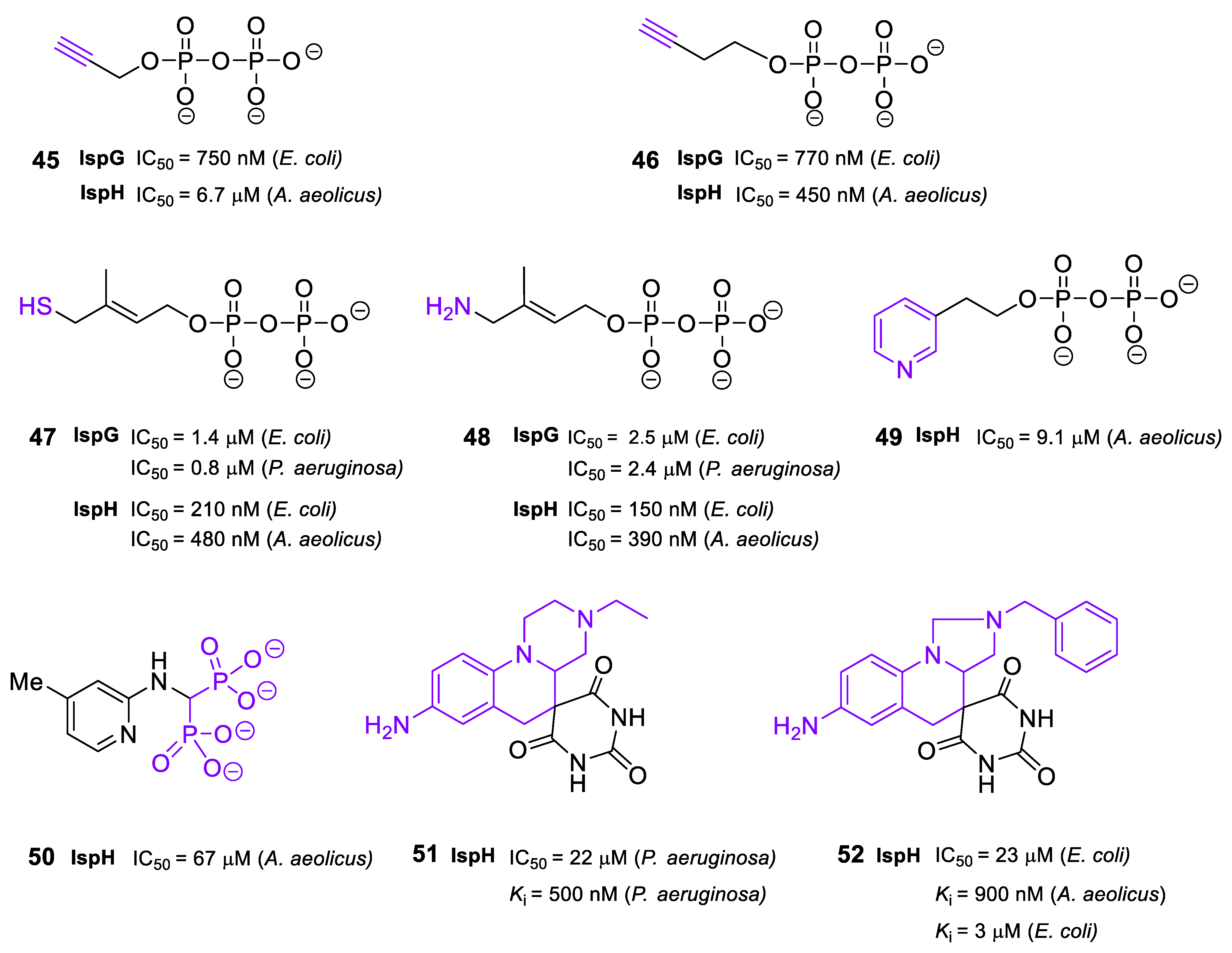
2.6. Inhibitors of IspG and IspH/LytB
3. MEP Pathway as Immunostimulant Source
3.1. Natural Phosphoantigens from the Isoprenoid Biosynthetic Pathways
3.2. Synthetic HMBPP Analogs
3.3. Prodrugs of Phosphoantigens
4. MEP Pathway as a Potential Contributor to Bacterial Cell–Cell Communication
4.1. Cell–Cell Communication Signal Molecules in Bacteria
4.2. Inter-Kingdom Communication: (S)-4,5-Dihydroxy-2,3-pentanedione and Its Derivatives
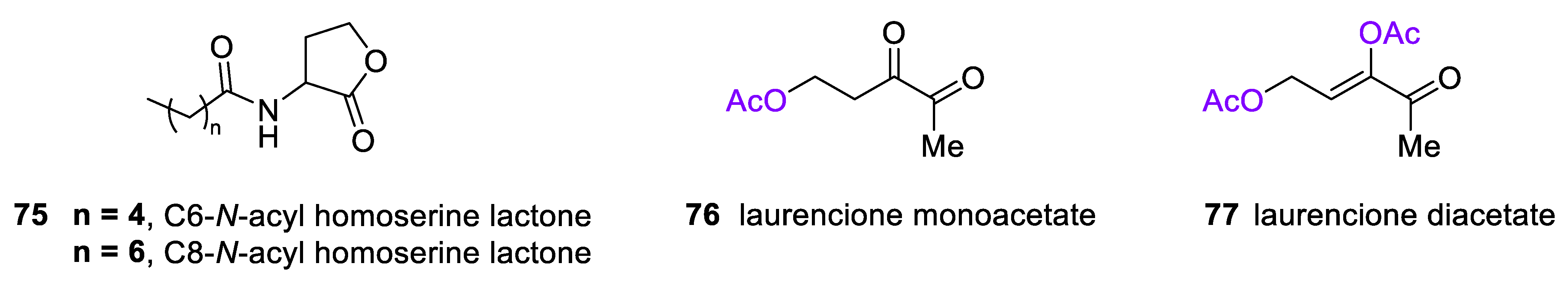
4.3. 4-Deoxy DPD (Laurencione), an MEP Pathway Byproduct
5. Conclusions and Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rohmer, M.; Knani, M.; Simonin, P.; Sutter, B.; Sahm, H. Isoprenoid biosynthesis in bacteria: A novel pathway for the early steps leading to isopentenyl diphosphate. Biochem. J. 1993, 295, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of Life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Rohmer, M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep. 1999, 16, 565–574. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.; Groll, M. The methylerythritol phosphate pathway to isoprenoids. Chem. Rev. 2017, 117, 5675–5703. [Google Scholar] [CrossRef]
- Cane, D.E.; Du, S.; Robinson, J.K.; Hsiung, Y.; Spenser, I.D. Biosynthesis of vitamin B6: Enzymatic conversion of 1-deoxy-ᴅ-xylulose-5-phosphate to pyridoxol phosphate. J. Am. Chem. Soc. 1999, 121, 7722–7723. [Google Scholar] [CrossRef]
- Fitzpatrick, T.B.; Moccand, C.; Roux, C. Vitamin B6 biosynthesis: Charting the mechanistic landscape. ChemBioChem 2010, 11, 1185–1193. [Google Scholar] [CrossRef]
- David, S.; Estramareix, B.; Fischer, J.C.; Therisod, M. 1-Deoxy-ᴅ-threo-2-pentulose: The precursor of the five-carbon chain of the thiazole of thiamine. J. Am. Chem. Soc. 1981, 103, 7341–7342. [Google Scholar] [CrossRef]
- Park, J.-H.; Dorrestein, P.C.; Zhai, H.; Kinsland, C.; McLafferty, F.W.; Begley, T.P. Biosynthesis of the thiazole moiety of thiamin pyrophosphate (Vitamin B1). Biochemistry 2003, 42, 12430–12438. [Google Scholar] [CrossRef]
- Masini, T.; Hirsch, A.K.H. Development of inhibitors of the 2-C-methyl-ᴅ-erythritol 4-phosphate (MEP) pathway enzymes as potential anti-infective agents. J. Med. Chem. 2014, 57, 9740–9763. [Google Scholar] [CrossRef]
- Altincicek, B.; Moll, J.; Campos, N.; Foerster, G.; Beck, E.; Hoeffler, J.-F.; Grosdemange-Billiard, C.; Rodríguez-Concepción, M.; Rohmer, M.; Boronat, A.; et al. Cutting Edge: Human γδ T cells are activated by intermediates of the 2-C-methyl-ᴅ-erythritol 4-phosphate pathway of isoprenoid biosynthesis. J. Immunol. 2001, 166, 3655–3658. [Google Scholar] [CrossRef] [Green Version]
- Rosa Putra, S.; Charon, L.; Danielsen, K.; Pale-Grosdemange, C.; Lois, L.-M.; Campos, N.; Boronat, A.; Rohmer, M. 5-Hydroxypentane-2,3-dione (laurencione), a bacterial metabolite of 1-deoxy-ᴅ-threo-pentulose. Tetrahedron Lett. 1998, 39, 6185–6188. [Google Scholar] [CrossRef]
- Hardie, K.R.; Heurlier, K. Establishing bacterial communities by “word of mouth”: LuxS and autoinducer 2 in biofilm development. Nat. Rev. Microbiol. 2008, 6, 635–643. [Google Scholar] [CrossRef]
- WHO Global Shortage of Innovative Antibiotics Fuels Emergence and Spread of Drug-Resistance. Available online: https://www.who.int/news/item/15-04-2021-global-shortage-of-innovative-antibiotics-fuels-emergence-and-spread-of-drug-resistance (accessed on 15 December 2022).
- Xiang, S.; Usunow, G.; Lange, G.; Busch, M.; Tong, L. Crystal structure of 1-deoxy-ᴅ-xylulose 5-phosphate synthase, a crucial enzyme for isoprenoids biosynthesis. J. Biol. Chem. 2007, 282, 2676–2682. [Google Scholar] [CrossRef]
- Bartee, D.; Freel Meyers, C.L. Toward understanding the chemistry and biology of 1-deoxy-ᴅ-xylulose 5-phosphate (DXP) synthase: A unique antimicrobial target at the heart of bacterial metabolism. Acc. Chem. Res. 2018, 51, 2546–2555. [Google Scholar] [CrossRef]
- Brammer Basta, L.A.; Patel, H.; Kakalis, L.; Jordan, F.; Freel Meyers, C.L. Defining critical residues for substrate binding to 1-deoxy-ᴅ-xylulose 5-phosphate synthase—active site substitutions stabilize the predecarboxylation intermediate C2α-lactylthiamin diphosphate. FEBS J. 2014, 281, 2820–2837. [Google Scholar] [CrossRef]
- Matsue, Y.; Mizuno, H.; Tomita, T.; Asami, T.; Nishiyama, M.; Kuzuyama, T. The herbicide ketoclomazone inhibits 1-deoxy-ᴅ-xylulose 5-phosphate synthase in the 2-C-methyl-ᴅ-erythritol 4-phosphate pathway and shows antibacterial activity against Haemophilus influenzae. J. Antibiot. (Tokyo) 2010, 63, 583–588. [Google Scholar] [CrossRef]
- Hayashi, D.; Kato, N.; Kuzuyama, T.; Sato, Y.; Ohkanda, J. Antimicrobial N-(2-chlorobenzyl)-substituted hydroxamate is an inhibitor of 1-deoxy-ᴅ-xylulose 5-phosphate synthase. Chem. Commun. 2013, 49, 5535. [Google Scholar] [CrossRef]
- Bartee, D.; Morris, F.; Al-khouja, A.; Freel Meyers, C.L. Hydroxybenzaldoximes are ᴅ-GAP-competitive inhibitors of E. coli 1-deoxy-ᴅ-xylulose-5-phosphate synthase. ChemBioChem 2015, 16, 1771–1781. [Google Scholar] [CrossRef]
- Smith, J.M.; Vierling, R.J.; Meyers, C.F. Selective inhibition of E. coli 1-deoxy-ᴅ-xylulose-5-phosphate synthase by acetylphosphonates. Med Chem Commun 2012, 3, 65–67. [Google Scholar] [CrossRef]
- Smith, J.M.; Warrington, N.V.; Vierling, R.J.; Kuhn, M.L.; Anderson, W.F.; Koppisch, A.T.; Freel Meyers, C.L. Targeting DXP synthase in human pathogens: Enzyme inhibition and antimicrobial activity of butylacetylphosphonate. J. Antibiot. (Tokyo) 2014, 67, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Bartee, D.; Freel Meyers, C.L. Targeting the unique mechanism of bacterial 1-deoxy-ᴅ-xylulose-5-phosphate synthase. Biochemistry 2018, 57, 4349–4356. [Google Scholar] [CrossRef] [PubMed]
- Bartee, D.; Sanders, S.; Phillips, P.D.; Harrison, M.J.; Koppisch, A.T.; Freel Meyers, C.L. Enamide prodrugs of acetyl phosphonate deoxy-ᴅ-xylulose-5-phosphate synthase inhibitors as potent antibacterial agents. ACS Infect. Dis. 2019, 5, 406–417. [Google Scholar] [CrossRef] [PubMed]
- Masini, T.; Lacy, B.; Monjas, L.; Hawksley, D.; de Voogd, A.R.; Illarionov, B.; Iqbal, A.; Leeper, F.J.; Fischer, M.; Kontoyianni, M.; et al. Validation of a homology model of Mycobacterium tuberculosis DXS: Rationalization of observed activities of thiamine derivatives as potent inhibitors of two orthologues of DXS. Org. Biomol. Chem. 2015, 13, 11263–11277. [Google Scholar] [CrossRef] [PubMed]
- Kuzuyama, T.; Shimizu, T.; Takahashi, S.; Seto, H. Fosmidomycin, a specific inhibitor of 1-deoxy-ᴅ-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway for terpenoid biosynthesis. Tetrahedron Lett. 1998, 39, 7913–7916. [Google Scholar] [CrossRef]
- Kuntz, L.; Tritsch, D.; Grosdemange-Billiard, C.; Hemmerlin, A.; Willem, A.; Bach, T.J.; Rohmer, M. Isoprenoid biosynthesis as a target for antibacterial and antiparasitic drugs: Phosphonohydroxamic acids as inhibitors of deoxyxylulose phosphate reducto-isomerase. Biochem. J. 2005, 386, 127–135. [Google Scholar] [CrossRef]
- Munier, M.; Tritsch, D.; Lièvremont, D.; Rohmer, M.; Grosdemange-Billiard, C. Synthesis and biological evaluation of aryl phosphoramidate prodrugs of fosfoxacin and its derivatives. Bioorganic Chem. 2019, 89, 103012. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Furukawa, S.; Ogihara, H.; Yamasaki, M. Fosmidomycin resistance in adenylate cyclase deficient (cya) mutants of Escherichia coli. Biosci. Biotechnol. Biochem. 2003, 67, 2030–2033. [Google Scholar] [CrossRef]
- Dhiman, R.K.; Schaeffer, M.L.; Bailey, A.M.; Testa, C.A.; Scherman, H.; Crick, D.C. 1-Deoxy-ᴅ-xylulose 5-phosphate reductoisomerase (IspC) from Mycobacterium tuberculosis: Towards understanding mycobacterial resistance to fosmidomycin. J. Bacteriol. 2005, 187, 8395–8402. [Google Scholar] [CrossRef]
- Na-Bangchang, K.; Ruengweerayut, R.; Karbwang, J.; Chauemung, A.; Hutchinson, D. Pharmacokinetics and pharmacodynamics of fosmidomycin monotherapy and combination therapy with clindamycin in the treatment of multidrug resistant falciparum malaria. Malar. J. 2007, 6, 70. [Google Scholar] [CrossRef]
- Piselli, C.; Benz, R. Fosmidomycin transport through the phosphate-specific porins OprO and OprP of Pseudomonas aeruginosa. Mol. Microbiol. 2021, 116, 97–108. [Google Scholar] [CrossRef]
- Katayama, N.; Tsubotani, S.; Nozaki, Y.; Harada, S.; Ono, H. Fosfadecin and fosfocytocin, new nucleotide antibiotics produced by bacteria. J. Antibiot. (Tokyo) 1990, 43, 238–246. [Google Scholar] [CrossRef]
- Woo, Y.-H.; Fernandes, R.P.M.; Proteau, P.J. Evaluation of fosmidomycin analogs as inhibitors of the Synechocystis sp. PCC6803 1-deoxy-ᴅ-xylulose 5-phosphate reductoisomerase. Bioorg. Med. Chem. 2006, 14, 2375–2385. [Google Scholar] [CrossRef]
- Munier, M. Synthèse de Prodrogues d’inhibiteurs de la 1-désoxy-ᴅ-xylulose 5-phosphate Réductoisomérase (DXR) : Des Agents Antituberculeux Potentiels. Ph.D. Thesis, Université de Strasbourg, Strasbourg, France, 2016. [Google Scholar]
- Jackson, E.R.; San Jose, G.; Brothers, R.C.; Edelstein, E.K.; Sheldon, Z.; Haymond, A.; Johny, C.; Boshoff, H.I.; Couch, R.D.; Dowd, C.S. The effect of chain length and unsaturation on Mtb Dxr inhibition and antitubercular killing activity of FR900098 analogs. Bioorg. Med. Chem. Lett. 2014, 24, 649–653. [Google Scholar] [CrossRef]
- Chofor, R.; Risseeuw, M.; Pouyez, J.; Johny, C.; Wouters, J.; Dowd, C.; Couch, R.; Van Calenbergh, S. Synthetic fosmidomycin analogues with altered chelating moieties do not inhibit 1-deoxy-ᴅ-xylulose 5-phosphate reductoisomerase or Plasmodium falciparum growth in vitro. Molecules 2014, 19, 2571–2587. [Google Scholar] [CrossRef]
- Andaloussi, M.; Lindh, M.; Björkelid, C.; Suresh, S.; Wieckowska, A.; Iyer, H.; Karlén, A.; Larhed, M. Substitution of the phosphonic acid and hydroxamic acid functionalities of the DXR inhibitor FR900098: An attempt to improve the activity against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2011, 21, 5403–5407. [Google Scholar] [CrossRef]
- Ponaire, S.; Zinglé, C.; Tritsch, D.; Grosdemange-Billiard, C.; Rohmer, M. Growth inhibition of Mycobacterium smegmatis by prodrugs of deoxyxylulose phosphate reducto-isomerase inhibitors, promising anti-mycobacterial agents. Eur. J. Med. Chem. 2012, 51, 277–285. [Google Scholar] [CrossRef]
- Munier, M.; Tritsch, D.; Krebs, F.; Esque, J.; Hemmerlin, A.; Rohmer, M.; Stote, R.H.; Grosdemange-Billiard, C. Synthesis and biological evaluation of phosphate isosters of fosmidomycin and analogs as inhibitors of Escherichia coli and Mycobacterium smegmatis 1-deoxyxylulose 5-phosphate reductoisomerases. Bioorg. Med. Chem. 2017, 25, 684–689. [Google Scholar] [CrossRef]
- Dreneau, A.; Krebs, F.S.; Munier, M.; Ngov, C.; Tritsch, D.; Lièvremont, D.; Rohmer, M.; Grosdemange-Billiard, C. α,α-Difluorophosphonohydroxamic acid derivatives among the best antibacterial fosmidomycin analogues. Molecules 2021, 26, 5111. [Google Scholar] [CrossRef]
- Xue, J.; Diao, J.; Cai, G.; Deng, L.; Zheng, B.; Yao, Y.; Song, Y. Antimalarial and structural studies of pyridine-containing inhibitors of 1-deoxyxylulose-5-phosphate reductoisomerase. ACS Med. Chem. Lett. 2013, 4, 278–282. [Google Scholar] [CrossRef]
- Haemers, T.; Wiesner, J.; Poecke, S.V.; Goeman, J.; Henschker, D.; Beck, E.; Jomaa, H.; Calenbergh, S.V. Synthesis of α-substituted fosmidomycin analogues as highly potent Plasmodium falciparum growth inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 1888–1891. [Google Scholar] [CrossRef] [Green Version]
- Kunfermann, A.; Lienau, C.; Illarionov, B.; Held, J.; Gräwert, T.; Behrendt, C.T.; Werner, P.; Hähn, S.; Eisenreich, W.; Riederer, U.; et al. IspC as target for antiinfective drug discovery: Synthesis, enantiomeric separation, and structural biology of fosmidomycin thia isosters. J. Med. Chem. 2013, 56, 8151–8162. [Google Scholar] [CrossRef] [PubMed]
- Ball, H.S.; Girma, M.; Zainab, M.; Riley, H.; Behrendt, C.T.; Lienau, C.; Konzuch, S.; Avelar, L.A.A.; Lungerich, B.; Soojhawon, I.; et al. Inhibition of the Yersinia pestis methylerythritol phosphate pathway of isoprenoid biosynthesis by α-phenyl-substituted reverse fosmidomycin analogues. ACS Omega 2020, 5, 5170–5175. [Google Scholar] [CrossRef] [PubMed]
- San Jose, G.; Jackson, E.R.; Haymond, A.; Johny, C.; Edwards, R.L.; Wang, X.; Brothers, R.C.; Edelstein, E.K.; Odom, A.R.; Boshoff, H.I.; et al. Structure–activity relationships of the MEPicides: N-Acyl and O-Linked analogs of FR900098 as inhibitors of Dxr from Mycobacterium tuberculosis and Yersinia pestis. ACS Infect. Dis. 2016, 2, 923–935. [Google Scholar] [CrossRef] [PubMed]
- McKenney, E.S.; Sargent, M.; Khan, H.; Uh, E.; Jackson, E.R.; Jose, G.S.; Couch, R.D.; Dowd, C.S.; van Hoek, M.L. Lipophilic prodrugs of FR900098 are antimicrobial against Francisella novicida in vivo and in vitro and show GlpT independent efficacy. PLoS ONE 2012, 7, e38167. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.C.; Parish, T. Dxr is essential in Mycobacterium tuberculosis and fosmidomycin resistance is due to a lack of uptake. BMC Microbiol. 2008, 8, 78. [Google Scholar] [CrossRef]
- Uh, E.; Jackson, E.R.; San Jose, G.; Maddox, M.; Lee, R.E.; Lee, R.E.; Boshoff, H.I.; Dowd, C.S. Antibacterial and antitubercular activity of fosmidomycin, FR900098, and their lipophilic analogs. Bioorg. Med. Chem. Lett. 2011, 21, 6973–6976. [Google Scholar] [CrossRef]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Maes, L.; Martin, A.; Van Calenbergh, S. Amino acid based prodrugs of a fosmidomycin surrogate as antimalarial and antitubercular agents. Bioorg. Med. Chem. 2019, 27, 729–747. [Google Scholar] [CrossRef]
- Courtens, C.; Risseeuw, M.; Caljon, G.; Maes, L.; Cos, P.; Martin, A.; Van Calenbergh, S. Double prodrugs of a fosmidomycin surrogate as antimalarial and antitubercular agents. Bioorg. Med. Chem. Lett. 2019, 29, 1232–1235. [Google Scholar] [CrossRef]
- Kuzuyama, T.; Takagi†, M.; Kaneda, K.; Dairi, T.; Seto, H. Formation of 4-(cytidine 5′-diphospho)-2-C-methyl-ᴅ-erythritol from 2-C-methyl-ᴅ-erythritol 4-phosphate by 2-C-methyl-ᴅ-erythritol 4-phosphate cytidylyltransferase, a new enzyme in the nonmevalonate pathway. Tetrahedron Lett. 2000, 41, 703–706. [Google Scholar] [CrossRef]
- Rohdich, F.; Wungsintaweekul, J.; Fellermeier, M.; Sagner, S.; Herz, S.; Kis, K.; Eisenreich, W.; Bacher, A.; Zenk, M.H. Cytidine 5′-triphosphate-dependent biosynthesis of isoprenoids: YgbP protein of Escherichia coli catalyzes the formation of 4-diphosphocytidyl-2-C-methylerythritol. Proc. Natl. Acad. Sci. USA 1999, 96, 11758–11763. [Google Scholar] [CrossRef] [Green Version]
- Schwab, A.; Illarionov, B.; Frank, A.; Kunfermann, A.; Seet, M.; Bacher, A.; Witschel, M.C.; Fischer, M.; Groll, M.; Diederich, F. Mechanism of allosteric inhibition of the enzyme IspD by three different classes of ligands. ACS Chem. Biol. 2017, 12, 2132–2138. [Google Scholar] [CrossRef]
- Riemersma, M.; Froese, D.S.; van Tol, W.; Engelke, U.F.; Kopec, J.; van Scherpenzeel, M.; Ashikov, A.; Krojer, T.; von Delft, F.; Tessari, M.; et al. Human ISPD is a cytidyltransferase required for dystroglycan O-mannosylation. Chem. Biol. 2015, 22, 1643–1652. [Google Scholar] [CrossRef]
- Bavin, E.M.; Kay, E.; Simmonite, D. Some observations on the activity of mixtures of antibacterial substances. J. Pharm. Pharmacol. 2011, 7, 676–682. [Google Scholar] [CrossRef]
- Gao, P.; Yang, Y.; Xiao, C.; Liu, Y.; Gan, M.; Guan, Y.; Hao, X.; Meng, J.; Zhou, S.; Chen, X.; et al. Identification and validation of a novel lead compound targeting 4-diphosphocytidyl-2-C-methylerythritol synthetase (IspD) of mycobacteria. Eur. J. Pharmacol. 2012, 694, 45–52. [Google Scholar] [CrossRef]
- Bartee, D.; Wheadon, M.J.; Freel Meyers, C.L. Synthesis and evaluation of fluoroalkyl phosphonyl analogues of 2-C-methylerythritol phosphate as substrates and inhibitors of IspD from human pathogens. J. Org. Chem. 2018, 83, 9580–9591. [Google Scholar] [CrossRef]
- Lillo, A.M.; Tetzlaff, C.N.; Sangari, F.J.; Cane, D.E. Functional expression and characterization of eryA, the erythritol kinase of Brucella abortus, and enzymatic synthesis of l-erythritol-4-phosphate. Bioorg. Med. Chem. Lett. 2003, 13, 737–739. [Google Scholar] [CrossRef]
- Ershov, Y.V. 2-C-methylerythritol phosphate pathway of isoprenoid biosynthesis as a target in identifying new antibiotics, herbicides, and immunomodulators: A review. Appl. Biochem. Microbiol. 2007, 43, 115–138. [Google Scholar] [CrossRef]
- Baatarkhuu, Z.; Chaignon, P.; Borel, F.; Ferrer, J.-L.; Wagner, A.; Seemann, M. Synthesis and kinetic evaluation of an azido analogue of methylerythritol phosphate: A novel inhibitor of E. coli YgbP/IspD. Sci. Rep. 2018, 8, 17892. [Google Scholar] [CrossRef]
- Zhang, B.; Watts, K.M.; Hodge, D.; Kemp, L.M.; Hunstad, D.A.; Hicks, L.M.; Odom, A.R. A second target of the antimalarial and antibacterial agent fosmidomycin revealed by cellular metabolic profiling. Biochemistry 2011, 50, 3570–3577. [Google Scholar] [CrossRef]
- Eoh, H.; Narayanasamy, P.; Brown, A.C.; Parish, T.; Brennan, P.J.; Crick, D.C. Expression and characterization of soluble 4-diphosphocytidyl-2-C-methyl-ᴅ-erythritol kinase from bacterial pathogens. Chem. Biol. 2009, 16, 1230–1239. [Google Scholar] [CrossRef] [Green Version]
- Sgraja, T.; Alphey, M.S.; Ghilagaber, S.; Marquez, R.; Robertson, M.N.; Hemmings, J.L.; Lauw, S.; Rohdich, F.; Bacher, A.; Eisenreich, W.; et al. Characterization of Aquifex aeolicus 4-diphosphocytidyl-2-C-methyl-ᴅ-erythritol kinase—ligand recognition in a template for antimicrobial drug discovery: Aquifex aeolicus IspE. FEBS J. 2008, 275, 2779–2794. [Google Scholar] [CrossRef] [PubMed]
- Schütz, A.P.; Locher, S.; Bernet, B.; Illarionov, B.; Fischer, M.; Bacher, A.; Diederich, F. 5-Substituted (1-thiolan-2-yl)cytosines as inhibitors of A. aeolicus and E. coli IspE kinases: Very different affinities to similar substrate-binding sites: Inhibitors of A. aeolicus and E. coli IspE kinases. Eur. J. Org. Chem. 2013, 2013, 880–887. [Google Scholar] [CrossRef]
- Hirsch, A.K.H.; Lauw, S.; Gersbach, P.; Schweizer, W.B.; Rohdich, F.; Eisenreich, W.; Bacher, A.; Diederich, F. Nonphosphate inhibitors of IspE protein, a kinase in the non-mevalonate pathway for isoprenoid biosynthesis and a potential target for antimalarial therapy. ChemMedChem 2007, 2, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.K.H.; Diederich, F.; Antonietti, M.; Börner, H.G. Bioconjugates to specifically render inhibitors water-soluble. Soft Matter 2010, 6, 88–91. [Google Scholar] [CrossRef]
- Tang, M.; Odejinmi, S.I.; Allette, Y.M.; Vankayalapati, H.; Lai, K. Identification of novel small molecule inhibitors of 4-diphosphocytidyl-2-C-methyl-ᴅ-erythritol (CDP-ME) kinase of Gram-negative bacteria. Bioorg. Med. Chem. 2011, 19, 5886–5895. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, S.; Kaiser, J.; Wungsintaweekul, J.; Hecht, S.; Eisenreich, W.; Gerhardt, S.; Bacher, A.; Rohdich, F. Structure of 2-C-methyl-ᴅ-erythritol-2,4-cyclodiphosphate synthase involved in mevalonate-independent biosynthesis of isoprenoids. J. Mol. Biol. 2002, 316, 79–88. [Google Scholar] [CrossRef]
- Kemp, L.E.; Alphey, M.S.; Bond, C.S.; Ferguson, M.A.J.; Hecht, S.; Bacher, A.; Eisenreich, W.; Rohdich, F.; Hunter, W.N. The identification of isoprenoids that bind in the intersubunit cavity of Escherichia coli 2-C-methyl-ᴅ-erythritol-2,4-cyclodiphosphate synthase by complementary biophysical methods. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 45–52. [Google Scholar] [CrossRef]
- Bitok, J.K.; Meyers, C.F. 2-C-Methyl-ᴅ-erythritol 4-phosphate enhances and sustains cyclodiphosphate synthase IspF activity. ACS Chem. Biol. 2012, 7, 1702–1710. [Google Scholar] [CrossRef]
- Kipchirchir Bitok, J.; Meyers, C.F. Synthesis and evaluation of stable substrate analogs as potential modulators of cyclodiphosphate synthase IspF. MedChemComm 2013, 4, 130–134. [Google Scholar] [CrossRef]
- Geist, J.G.; Lauw, S.; Illarionova, V.; Illarionov, B.; Fischer, M.; Gräwert, T.; Rohdich, F.; Eisenreich, W.; Kaiser, J.; Groll, M.; et al. Thiazolopyrimidine inhibitors of 2-methylerythritol 2,4-cyclodiphosphate synthase (IspF) from Mycobacterium tuberculosis and Plasmodium falciparum. ChemMedChem 2010, 5, 1092–1101. [Google Scholar] [CrossRef]
- Thelemann, J.; Illarionov, B.; Barylyuk, K.; Geist, J.; Kirchmair, J.; Schneider, P.; Anthore, L.; Root, K.; Trapp, N.; Bacher, A.; et al. Aryl bis-sulfonamide inhibitors of IspF from Arabidopsis thaliana and Plasmodium falciparum. ChemMedChem 2015, 10, 2090–2098. [Google Scholar] [CrossRef]
- Root, K.; Barylyuk, K.; Schwab, A.; Thelemann, J.; Illarionov, B.; Geist, J.G.; Gräwert, T.; Bacher, A.; Fischer, M.; Diederich, F.; et al. Aryl bis-sulfonamides bind to the active site of a homotrimeric isoprenoid biosynthesis enzyme IspF and extract the essential divalent metal cation cofactor. Chem. Sci. 2018, 9, 5976–5986. [Google Scholar] [CrossRef]
- Liu, Z.; Jin, Y.; Liu, W.; Tao, Y.; Wang, G. Crystal structure of IspF from Bacillus subtilis and absence of protein complex assembly amongst IspD/IspE/IspF enzymes in the MEP pathway. Biosci. Rep. 2018, 38, BSR20171370. [Google Scholar] [CrossRef]
- Honold, A.; Lettl, C.; Schindele, F.; Illarionov, B.; Haas, R.; Witschel, M.; Bacher, A.; Fischer, M. Inhibitors of the bifunctional 2-C-methyl-d-erythritol 4-phosphate cytidylyl transferase/2-C-methyl-ᴅ-erythritol-2,4-cyclopyrophosphate synthase (IspDF) of Helicobacter pylori. Helv. Chim. Acta 2019, 102, e1800228. [Google Scholar] [CrossRef]
- Quitterer, F.; Frank, A.; Wang, K.; Rao, G.; O’Dowd, B.; Li, J.; Guerra, F.; Abdel-Azeim, S.; Bacher, A.; Eppinger, J.; et al. Atomic-resolution structures of discrete stages on the reaction coordinate of the [Fe4-S4] enzyme IspG (GcpE). J. Mol. Biol. 2015, 427, 2220–2228. [Google Scholar] [CrossRef]
- Rekittke, I.; Jomaa, H.; Ermler, U. Structure of the GcpE (IspG)-MEcPP complex from Thermus thermophilus. FEBS Lett. 2012, 586, 3452–3457. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Guerra, F.; Wang, K.; Wang, W.; Li, J.; Huang, C.; Zhu, W.; Houlihan, K.; Li, Z.; Zhang, Y.; et al. Structure, function and inhibition of the two- and three-domain 4Fe-4S IspG proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 8558–8563. [Google Scholar] [CrossRef]
- Wang, W.; Li, J.; Wang, K.; Huang, C.; Zhang, Y.; Oldfield, E. Organometallic mechanism of action and inhibition of the 4Fe-4S isoprenoid biosynthesis protein GcpE (IspG). Proc. Natl. Acad. Sci. USA 2010, 107, 11189–11193. [Google Scholar] [CrossRef]
- Lee, M.; Gräwert, T.; Quitterer, F.; Rohdich, F.; Eppinger, J.; Eisenreich, W.; Bacher, A.; Groll, M. Biosynthesis of isoprenoids: Crystal structure of the [4Fe-4S] cluster protein IspG. J. Mol. Biol. 2010, 404, 600–610. [Google Scholar] [CrossRef]
- Masini, T.; Kroezen, B.S.; Hirsch, A.K.H. Druggability of the enzymes of the non-mevalonate pathway. Drug Discov. Today 2013, 18, 1256–1262. [Google Scholar] [CrossRef]
- Janthawornpong, K.; Krasutsky, S.; Chaignon, P.; Rohmer, M.; Poulter, C.D.; Seemann, M. Inhibition of IspH, a [4Fe-4S]2+ enzyme involved in the biosynthesis of isoprenoids via the methylerythritol phosphate pathway. J. Am. Chem. Soc. 2013, 135, 1816–1822. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Wang, K.; Li, J.; Wang, W.; Liu, Y.-L.; Amin, S.; Oldfield, E. Inhibition of the 4Fe–4S proteins IspG and IspH: An EPR, ENDOR and HYSCORE investigation. Chem. Sci. 2014, 5, 1642–1649. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Wang, W.; No, J.-H.; Zhang, Y.; Zhang, Y.; Oldfield, E. Inhibition of the Fe4S4-cluster-containing protein IspH (LytB): Electron Paramagnetic Resonance, metallacycles, and mechanisms. J. Am. Chem. Soc. 2010, 132, 6719–6727. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.; Williams, S.; Wang, H.; No, J.H.; Rao, G.; Wang, W.; McCammon, J.A.; Cramer, S.P.; Oldfield, E. Spectroscopic and computational investigations of ligand binding to IspH: Discovery of non-diphosphate inhibitors. ChemBioChem 2017, 18, 914–920. [Google Scholar] [CrossRef]
- Tanaka, Y.; Sano, S.; Nieves, E.; De Libero, G.; Rosa, D.; Modlin, R.L.; Brenner, M.B.; Bloom, B.R.; Morita, C.T. Nonpeptide ligands for human gamma delta T cells. Proc. Natl. Acad. Sci. USA 1994, 91, 8175–8179. [Google Scholar] [CrossRef]
- Dimova, T.; Brouwer, M.; Gosselin, F.; Tassignon, J.; Leo, O.; Donner, C.; Marchant, A.; Vermijlen, D. Effector Vγ9Vδ2 T cells dominate the human fetal γδ T-cell repertoire. Proc. Natl. Acad. Sci. USA 2015, 112, E556–E565. [Google Scholar] [CrossRef]
- Herrmann, T.; Karunakaran, M.M.; Fichtner, A.S. A glance over the fence: Using phylogeny and species comparison for a better understanding of antigen recognition by human γδ T-cells. Immunol. Rev. 2020, 298, 218–236. [Google Scholar] [CrossRef]
- Bonneville, M.; O’Brien, R.L.; Born, W.K. γδ T cell effector functions: A blend of innate programming and acquired plasticity. Nat. Rev. Immunol. 2010, 10, 467–478. [Google Scholar] [CrossRef]
- Riganti, C.; Massaia, M.; Davey, M.S.; Eberl, M. Human γδ T-cell responses in infection and immunotherapy: Common mechanisms, common mediators? Highlights. Eur. J. Immunol. 2012, 42, 1668–1676. [Google Scholar] [CrossRef]
- Harly, C.; Peigné, C.-M.; Scotet, E. Molecules and mechanisms implicated in the peculiar antigenic activation process of human Vγ9Vδ2 T cells. Front. Immunol. 2015, 5, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Moritat, C.T.; Tanaka, Y.; Nievest, E.; Brennert, M.B.; Bloom, B.R. Natural and synthetic non-peptide antigens recognized by human γδ T cells. Nature 1995, 375, 155–158. [Google Scholar] [CrossRef]
- Espinosa, E.; Belmant, C.; Sicard, H.; Poupot, R.; Bonneville, M.; Fournié, J.-J. Y2K+1 state-of-the-art on non-peptide phosphoantigens, a novel category of immunostimulatory molecules. Microbes Infect. 2001, 3, 645–654. [Google Scholar] [CrossRef]
- Lentini, N.A.; Schroeder, C.M.; Harmon, N.M.; Huang, X.; Schladetsch, M.A.; Foust, B.J.; Poe, M.M.; Hsiao, C.-H.C.; Wiemer, A.J.; Wiemer, D.F. Synthesis and metabolism of BTN3A1 ligands: Studies on modifications of the allylic alcohol. ACS Med. Chem. Lett. 2021, 12, 136–142. [Google Scholar] [CrossRef]
- Gober, H.-J.; Kistowska, M.; Angman, L.; Jenö, P.; Mori, L.; De Libero, G. Human T cell receptor γδ cells recognize endogenous mevalonate metabolites in tumor cells. J. Exp. Med. 2003, 197, 163–168. [Google Scholar] [CrossRef]
- Kunzmann, V.; Bauer, E.; Feurle, J.; Tony, F.W.H.P.; Wilhelm, M. Stimulation of γδ T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma. Blood 2000, 96, 384–392. [Google Scholar] [CrossRef]
- Wiemer, A.J. Structure-activity relationships of butyrophilin 3 ligands. ChemMedChem 2020, 15, 1030–1039. [Google Scholar] [CrossRef]
- Behr, C.; Poupot, R.; Peyrat, M.-A.; Poquet, Y.; Constant, P.; Dubois, P.; Bonneville, M.; Fournie, J.-J. Plasmodium falciparum stimuli for human γδ T cells are related to phosphorylated antigens of mycobacteria. Infect. Immun. 1996, 64, 2892–2896. [Google Scholar] [CrossRef]
- Hintz, M.; Reichenberg, A.; Altincicek, B.; Bahr, U.; Gschwind, R.M.; Kollas, A.-K.; Beck, E.; Wiesner, J.; Eberl, M.; Jomaa, H. Identification of (E)-4-hydroxy-3-methyl-but-2-enyl pyrophosphate as a major activator for human γδ T cells in Escherichia coli. FEBS Lett. 2001, 509, 317–322. [Google Scholar] [CrossRef]
- Eberl, M.; Altincicek, B.; Kollas, A.-K.; Sanderbrand, S.; Bahr, U.; Reichenberg, A.; Beck, E.; Foster, D.; Wiesner, J.; Hintz, M.; et al. Accumulation of a potent γδ T-cell stimulator after deletion of the lytB gene in Escherichia coli. Immunology 2002, 106, 200–211. [Google Scholar] [CrossRef]
- Eberl, M.; Hintz, M.; Reichenberg, A.; Kollas, A.-K.; Wiesner, J.; Jomaa, H. Microbial isoprenoid biosynthesis and human γδ T cell activation. FEBS Lett. 2003, 544, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, E.; Belmant, C.; Pont, F.; Luciani, B.; Poupot, R.; Romagné, F.; Brailly, H.; Bonneville, M.; Fournié, J.-J. Chemical synthesis and biological activity of bromohydrin pyrophosphate, a potent stimulator of human γδ T cells. J. Biol. Chem. 2001, 276, 18337–18344. [Google Scholar] [CrossRef] [PubMed]
- Belmant, C.; Espinosa, E.; Halary, F.; Tang, Y.; Peyrat, M.-A.; Sicard, H.; Kozikowski, A.; Buelow, R.; Poupot, R.; Bonneville, M.; et al. A chemical basis for recognition of nonpeptide antigens by human γδ T cells. FASEB J. 2000, 14, 1669–1670. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, J.; Levy, V.; Sicard, H.; Senellart, H.; Audrain, M.; Hiret, S.; Rolland, F.; Bruzzoni-Giovanelli, H.; Rimbert, M.; Galéa, C.; et al. Phase I study of bromohydrin pyrophosphate (BrHPP, IPH 1101), a Vγ9Vδ2 T lymphocyte agonist in patients with solid tumors. Cancer Immunol. Immunother. 2010, 59, 1521–1530. [Google Scholar] [CrossRef] [PubMed]
- Fournié, J.-J.; Sicard, H.; Poupot, M.; Bezombes, C.; Blanc, A.; Romagné, F.; Ysebaert, L.; Laurent, G. What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell. Mol. Immunol. 2013, 10, 35–41. [Google Scholar] [CrossRef]
- Wiemer, D.F.; Wiemer, A.J. Opportunities and challenges in development of phosphoantigens as Vγ9Vδ2 T cell agonists. Biochem. Pharmacol. 2014, 89, 301–312. [Google Scholar] [CrossRef]
- Sicard, H.; Ingoure, S.; Luciani, B.; Serraz, C.; Fournié, J.-J.; Bonneville, M.; Tiollier, J.; Romagné, F. In vivo immunomanipulation of Vγ9Vδ2 T cells with a synthetic phosphoantigen in a preclinical nonhuman primate model. J. Immunol. 2005, 175, 5471–5480. [Google Scholar] [CrossRef]
- Boëdec, A.; Sicard, H.; Dessolin, J.; Herbette, G.; Ingoure, S.; Raymond, C.; Belmant, C.; Kraus, J.-L. Synthesis and biological activity of phosphonate analogues and geometric isomers of the highly potent phosphoantigen (E)-1-hydroxy-2-methylbut-2-enyl 4-diphosphate. J. Med. Chem. 2008, 51, 1747–1754. [Google Scholar] [CrossRef]
- Hsiao, C.-H.C.; Lin, X.; Barney, R.J.; Shippy, R.R.; Li, J.; Vinogradova, O.; Wiemer, D.F.; Wiemer, A.J. Synthesis of a phosphoantigen prodrug that potently activates Vγ9Vδ2 T-lymphocytes. Chem. Biol. 2014, 21, 945–954. [Google Scholar] [CrossRef]
- Shippy, R.R.; Lin, X.; Agabiti, S.S.; Li, J.; Zangari, B.M.; Foust, B.J.; Poe, M.M.; Hsiao, C.-H.C.; Vinogradova, O.; Wiemer, D.F.; et al. Phosphinophosphonates and their tris-pivaloyloxymethyl prodrugs reveal a negatively cooperative butyrophilin activation mechanism. J. Med. Chem. 2017, 60, 2373–2382. [Google Scholar] [CrossRef]
- Foust, B.J.; Poe, M.M.; Lentini, N.A.; Hsiao, C.-H.C.; Wiemer, A.J.; Wiemer, D.F. Mixed aryl phosphonate prodrugs of a butyrophilin ligand. ACS Med. Chem. Lett. 2017, 8, 914–918. [Google Scholar] [CrossRef]
- Foust, B.J.; Li, J.; Hsiao, C.C.; Wiemer, D.F.; Wiemer, A.J. Stability and efficiency of mixed aryl phosphonate prodrugs. ChemMedChem 2019, 14, 1597–1603. [Google Scholar] [CrossRef]
- Davey, M.S.; Malde, R.; Mykura, R.C.; Baker, A.T.; Taher, T.E.; Le Duff, C.S.; Willcox, B.E.; Mehellou, Y. Synthesis and biological evaluation of (E)-4-hydroxy-3-methylbut-2-enyl phosphate (HMBP) aryloxy triester phosphoramidate prodrugs as activators of Vγ9/Vδ2 T-cell immune responses. J. Med. Chem. 2018, 61, 2111–2117. [Google Scholar] [CrossRef]
- Lentini, N.A.; Foust, B.J.; Hsiao, C.-H.C.; Wiemer, A.J.; Wiemer, D.F. Phosphonamidate prodrugs of a butyrophilin ligand display plasma stability and potent Vγ9 Vδ2 T cell stimulation. J. Med. Chem. 2018, 61, 8658–8669. [Google Scholar] [CrossRef]
- Kadri, H.; Taher, T.E.; Xu, Q.; Sharif, M.; Ashby, E.; Bryan, R.T.; Willcox, B.E.; Mehellou, Y. Aryloxy diester phosphonamidate prodrugs of phosphoantigens (ProPAgens) as potent activators of Vγ9/Vδ2 T-cell immune responses. J. Med. Chem. 2020, 63, 11258–11270. [Google Scholar] [CrossRef]
- Harmon, N.M.; Huang, X.; Schladetsch, M.A.; Hsiao, C.-H.C.; Wiemer, A.J.; Wiemer, D.F. Potent double prodrug forms of synthetic phosphoantigens. Bioorg. Med. Chem. 2020, 28, 115666. [Google Scholar] [CrossRef]
- Harmon, N.M.; Poe, M.M.; Huang, X.; Singh, R.; Foust, B.J.; Hsiao, C.-H.C.; Wiemer, D.F.; Wiemer, A.J. Synthesis and metabolism of BTN3A1 ligands: Studies on diene modifications to the phosphoantigen scaffold. ACS Med. Chem. Lett. 2022, 13, 164–170. [Google Scholar] [CrossRef]
- Shapiro, J.; Dworkin, M. Bacteria as Multicellular Organisms; Oxford University Press: Oxford, UK, 1997; ISBN 978-0-19-509159-5. [Google Scholar]
- Mowat, E.; Rajendran, R.; Williams, C.; McCulloch, E.; Jones, B.; Lang, S.; Ramage, G. Pseudomonas aeruginosa and their small diffusible extracellular molecules inhibit Aspergillus fumigatus biofilm formation. FEMS Microbiol. Lett. 2010, 313, 96–102. [Google Scholar] [CrossRef]
- Schmidt, R.; Cordovez, V.; de Boer, W.; Raaijmakers, J.; Garbeva, P. Volatile affairs in microbial interactions. ISME J. 2015, 9, 2329–2335. [Google Scholar] [CrossRef]
- Briard, B.; Heddergott, C.; Latgé, J.-P. Volatile compounds emitted by Pseudomonas aeruginosa stimulate growth of the fungal pathogen Aspergillus fumigatus. mBio 2016, 7, e00219-16. [Google Scholar] [CrossRef]
- Prescott, R.D.; Decho, A.W. Flexibility and adaptability of quorum sensing in nature. Trends Microbiol. 2020, 28, 436–444. [Google Scholar] [CrossRef]
- Duerre, J.A.; Baker, D.J. Structure elucidation of a carbohydrate derived from S-ribosylhomocysteine by enzymatic cleavage. Fed. Proc. 1971, 30, 1067. [Google Scholar]
- Bassler, B.L.; Wright, M.; Silverman, M.R. Multiple signalling systems controlling expression of luminescence in Vibrio harveyi: Sequence and function of genes encoding a second sensory pathway. Mol. Microbiol. 1994, 13, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Xavier, K.B.; Bassler, B.L. LuxS quorum sensing: More than just a numbers game. Curr. Opin. Microbiol. 2003, 6, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Di Cagno, R.; De Angelis, M.; Calasso, M.; Gobbetti, M. Proteomics of the bacterial cross-talk by quorum sensing. J. Proteomics 2011, 74, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Lowery, C.A.; Park, J.; Kaufmann, G.F.; Janda, K.D. An unexpected switch in the modulation of AI-2-based quorum sensing discovered through synthetic 4,5-dihydroxy-2,3-pentanedione analogues. J. Am. Chem. Soc. 2008, 130, 9200–9201. [Google Scholar] [CrossRef]
- Duan, K.; Dammel, C.; Stein, J.; Rabin, H.; Surette, M.G. Modulation of Pseudomonas aeruginosa gene expression by host microflora through interspecies communication: Modulation of P. aeruginosa by oropharyngeal flora. Mol. Microbiol. 2003, 50, 1477–1491. [Google Scholar] [CrossRef]
- Li, H.; Li, X.; Wang, Z.; Fu, Y.; Ai, Q.; Dong, Y.; Yu, J. Autoinducer-2 regulates Pseudomonas aeruginosa PAO1 biofilm formation and virulence production in a dose-dependent manner. BMC Microbiol. 2015, 15, 192. [Google Scholar] [CrossRef]
- Roy, V.; Meyer, M.T.; Smith, J.A.I.; Gamby, S.; Sintim, H.O.; Ghodssi, R.; Bentley, W.E. AI-2 analogs and antibiotics: A synergistic approach to reduce bacterial biofilms. Appl. Microbiol. Biotechnol. 2013, 97, 2627–2638. [Google Scholar] [CrossRef]
- Kim, Y.; Oh, S.; Ahn, E.Y.; Imm, J.-Y.; Oh, S.; Park, S.; Kim, S.H. Proteome analysis of virulence factor regulated by autoinducer-2–like activity in Escherichia coli O157:H7. J. Food Prot. 2007, 70, 300–307. [Google Scholar] [CrossRef]
- Rohmer, M. The mevalonate-independent methylerythritol 4-phosphate (MEP) pathway for isoprenoid biosynthesis, including carotenoids. Pure Appl. Chem. 1999, 71, 2279–2284. [Google Scholar] [CrossRef]
- Bernart, M.W.; Gerwick, W.H.; Corcoran, E.E.; Lee, A.Y.; Clardy, J. Laurencione, a heterocycle from the red alga Laurencia spectabilis. Phytochemistry 1992, 31, 1273–1276. [Google Scholar] [CrossRef]
- Lowery, C.A.; McKenzie, K.M.; Qi, L.; Meijler, M.M.; Janda, K.D. Quorum sensing in Vibrio harveyi: Probing the specificity of the LuxP binding site. Bioorg. Med. Chem. Lett. 2005, 15, 2395–2398. [Google Scholar] [CrossRef]
- Gerwick, L.; Boudreau, P.; Choi, H.; Mascuch, S.; Villa, F.A.; Balunas, M.J.; Malloy, K.L.; Teasdale, M.E.; Rowley, D.C.; Gerwick, W.H. Interkingdom signaling by structurally related cyanobacterial and algal secondary metabolites. Phytochem. Rev. 2013, 12, 459–465. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Geller, L.T.; Straussman, R. Intratumoral bacteria may elicit chemoresistance by metabolizing anticancer agents. Mol. Cell. Oncol. 2018, 5, e1405139. [Google Scholar] [CrossRef] [Green Version]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allamand, A.; Piechowiak, T.; Lièvremont, D.; Rohmer, M.; Grosdemange-Billiard, C. The Multifaceted MEP Pathway: Towards New Therapeutic Perspectives. Molecules 2023, 28, 1403. https://doi.org/10.3390/molecules28031403
Allamand A, Piechowiak T, Lièvremont D, Rohmer M, Grosdemange-Billiard C. The Multifaceted MEP Pathway: Towards New Therapeutic Perspectives. Molecules. 2023; 28(3):1403. https://doi.org/10.3390/molecules28031403
Chicago/Turabian StyleAllamand, Alizée, Teresa Piechowiak, Didier Lièvremont, Michel Rohmer, and Catherine Grosdemange-Billiard. 2023. "The Multifaceted MEP Pathway: Towards New Therapeutic Perspectives" Molecules 28, no. 3: 1403. https://doi.org/10.3390/molecules28031403