
Identification of Phytochemicals in Bioactive Extracts of Acacia saligna Growing in Australia



Abstract
:1. Introduction
2. Results and Discussion
2.1. Fractionation of Crude Extracts of A. saligna
2.2. Antioxidant Evaluation of Fractions
2.2.1. DPPH Scavenging Activity
2.2.2. ABTS●+ Radical Assay
2.3. Inhibition of Yeast A-Glucosidase Enzyme Assay
2.4. Toxicity of Bioactive Methanolic Fractions against 3T3-L1 Adipocytes
2.5. Isolation of Pure Compounds Active Fractions
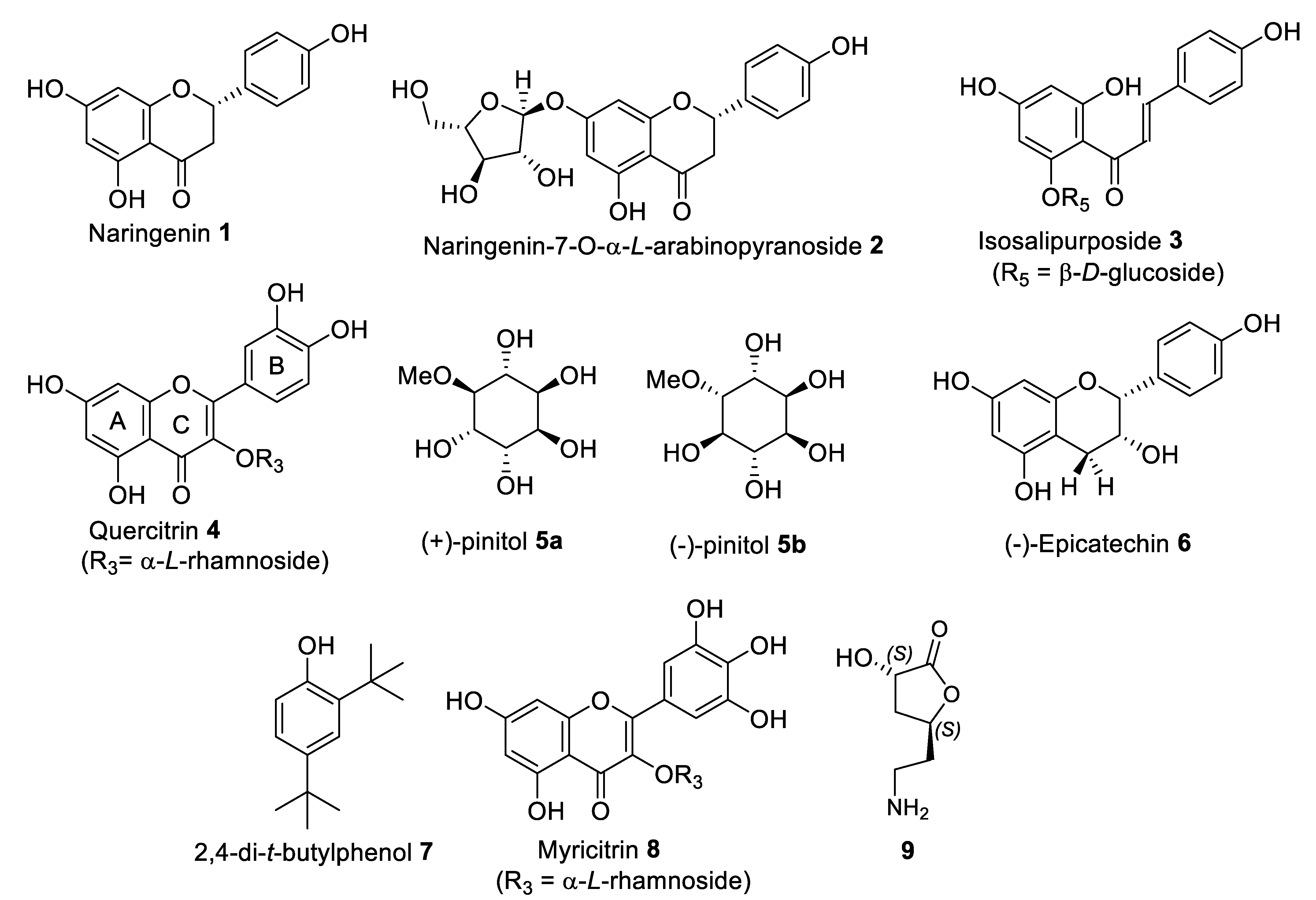
2.5.1. Isolation and Structural Identification of Compounds from FL-MeOH
2.5.2. Isolation and Structural Identification of Compounds from LF-MeOH
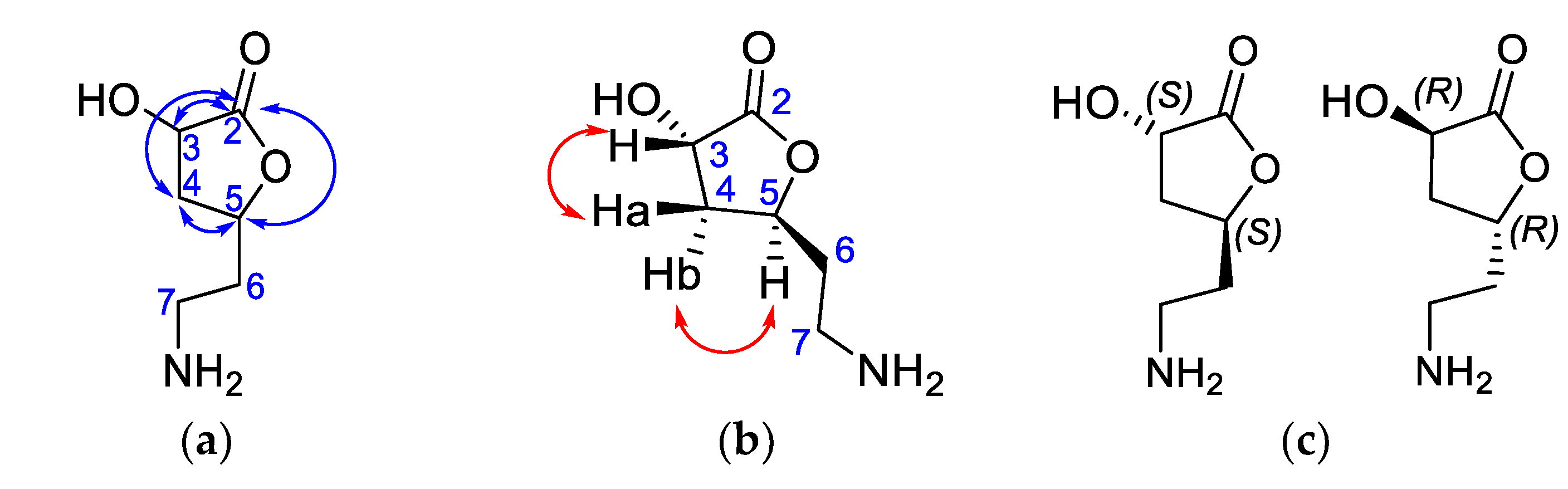
2.5.3. Isolation and Structural Identification of Compounds from BK-MeOH
2.6. Biological Activities of Isolated Compounds
2.6.1. Scavenging Activities of Isolates against DPPH and ABTS Free Radicals
2.6.2. α-Glucosidase Inhibition of Isolated Compounds
3. Materials and Methods
3.1. General Experimental for Phytochemical Analysis
3.2. Sample Collection and Identification
3.3. Sequential Extraction of Plant Parts
3.4. General Fractionation Method of Each Methanolic Extract of A. saligna
3.5. Fractionation of LF-MeOH
3.6. Fractionation of BK-MeOH
3.7. Spectral Data Analysis
3.8. Biological Assay
Materials
3.9. DPPH Scavenging Assay
3.10. ABTS●+ Radical Decolourisation Assay
3.11. In Vitro Assay of Yeast α-Glucosidase Inhibition
3.12. Differentiation of 3T3-L1 Preadipocytes into Adipocytes
3.13. Effects of Extracts on the Viability of 3T3-L1 Adipocytes
3.14. Statistical Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Orchard, A.E.; Wilson, A.G. Flora of Australia: Volume 11B, Mimosaceae, Acacia Part 2; CSIRO: Melbourne, Australia, 2001; p. 475. [Google Scholar]
- Ghribia, L.; Ghouilaa, H.; Omrib, A.; Besbesb, M.; Ben Janneta, H. Antioxidant and anti–acetylcholinesterase activities of extracts and secondary metabolites from Acacia cyanophylla. Asian Pac. J. Trop. Biomed. 2014, 4, S417–S423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Huqail, A.A.; Behiry, S.I.; Salem, M.Z.; Ali, H.M.; Siddiqui, M.H.; Salem, A.Z. Antifungal, antibacterial, and antioxidant activities of Acacia saligna (Labill.) HL Wendl. flower extract: HPLC analysis of phenolic and flavonoid compounds. Molecules 2019, 24, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elansary, H.O.; Szopa, A.; Kubica, P.; Ekiert, H.; Al-Mana, F.A.; Al-Yafrsi, M.A. Antioxidant and biological activities of Acacia saligna and Lawsonia inermis natural populations. Plants 2020, 9, 908. [Google Scholar] [CrossRef] [PubMed]
- El Ayeb-Zakhama, A.; Sakka-Rouis, L.; Bergaoui, A.; Flamini, G.; Jannet, H.B.; Harzallah-Skhiri, F. Chemical composition and allelopathic potential of essential oils obtained from Acacia cyanophylla Lindl cultivated in Tunisia. Chem. Biodivers. 2015, 12, 615–626. [Google Scholar] [CrossRef]
- Kaouthar, J.; Fakhfakh, L.M.; Siddique, K.H.; Hessini, K. Effect of Acacia saligna (Labill.) Wendl. extracts on seed germination and seedling performance of three native Mediterranean shrubs. Bot. Lett. 2022, 169, 51–60. [Google Scholar]
- El-Toumy, S.A.; Salib, J.; Mohamed, W.; Morsy, F. Phytochemical and antimicrobial studies on Acacia saligna leaves. Egypt J. Chem. 2010, 53, 705–717. [Google Scholar]
- Gedara, S.R.; Galala, A.A. New cytotoxic spirostane saponin and biflavonoid glycoside from the leaves of Acacia saligna (Labill.) HL Wendl. Nat. Prod. Res. 2014, 28, 324–329. [Google Scholar] [CrossRef]
- Gumgumjee, N.M.; Hajar, A.S. Antimicrobial efficacy of Acacia saligna (Labill.) HL Wendl. and Cordia sinensis Lam. leaves extracts against some pathogenic microorganisms. Int. J. Microbiol. Immunol. Res 2015, 3, 51–57. [Google Scholar]
- Salem, M.Z.M.; Mohamed, A.; Ali, H.; Al Farraj, D. Characterization of Phytoconstituents from Alcoholic Extracts of Four Woody Species and Their Potential Uses for Management of Six Fusarium oxysporum Isolates Identified from Some Plant Hosts. Plants 2021, 10, 1325. [Google Scholar] [CrossRef]
- Buttner, D.H.; Reddy, S.; Koekemoer, T.; van de Venter, M. An in vitro assessment of the potential antidiabetic activity and cytotoxic effects of ethanolic and aqueous extracts from three invasive Australian acacias. S. Afr. J. Bot. 2021, 141, 1–11. [Google Scholar] [CrossRef]
- Shaer, E. Utilization of Acacia saligna as livestock fodder in arid and semi-arid areas in Egypt. Cah. Options Mediterr. 2000, 45, 213–217. [Google Scholar]
- Mousa, M. Effect of feeding acacia as supplements on the nutrient digestion, growth performance, carcass traits and some blood constituents of Awassi lambs under the conditions of North Sinai. Asian J. Anim. Sci. 2011, 5, 102–117. [Google Scholar] [CrossRef] [Green Version]
- Gebru, G.; Tesfay, Y. Utilization of wheat bran and dried Acacia saligna (Labill) HL Wendl leaves by highland rams. Afr. J. Agric. Res. 2017, 12, 1286–1292. [Google Scholar]
- El-Toumy, S. Flavonoids from Acacia saligna leaves and Evaluation of Antihyperglycaemic Effect of Aqueous Extract. Planta Med. 2006, 72, P_004. [Google Scholar] [CrossRef]
- Leahy, J.L.; Hirsch, I.B.; Peterson, K.; Schneider, D.; Schneider, D. Targeting β-cell function early in the course of therapy for type 2 diabetes mellitus. J. Clin. Endocr. Metab. 2010, 95, 4206–4216. [Google Scholar] [CrossRef] [PubMed]
- Glastras, S.J.; Chen, H.; Teh, R.; McGrath, R.T.; Chen, J.; Pollock, C.A.; Wong, M.G.; Saad, S. Mouse models of diabetes, obesity and related kidney disease. PLoS ONE 2016, 11, e0162131–e0162146. [Google Scholar] [CrossRef] [Green Version]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Ghani, U. Re-exploring promising α-glucosidase inhibitors for potential development into oral anti-diabetic drugs: Finding needle in the haystack. Eur. J. Med. Chem. 2015, 103, 133–162. [Google Scholar] [CrossRef]
- Şöhretoğlu, D.; Sari, S. Flavonoids as alpha-glucosidase inhibitors: Mechanistic approaches merged with enzyme kinetics and molecular modelling. Phytochem. Rev. 2020, 19, 1081–1092. [Google Scholar] [CrossRef]
- Zreen, Z.; Hameed, A.; Kiran, S.; Farooq, T.; Zaroog, M.S. A Comparative Study of Diospyros malabarica (Gaub) Extracts in Various Polarity-Dependent Solvents for Evaluation of Phy-toconstituents and Biological Activities. BioMed Res. Int. 2022, 2022, 4746223. [Google Scholar] [CrossRef]
- Zohra, T.; Ovais, M.; Khalil, A.T.; Qasim, M.; Ayaz, M.; Shinwari, Z.K. Extraction optimisation, total phenolic, flavonoid contents, HPLC-DAD analysis and diverse pharmacological evaluations of Dysphania ambrosioides (L.) Mosyakin & Clemants. Nat. Prod. Res. 2019, 33, 136–142. [Google Scholar]
- Subhan, N. Phytochemical and Pharmacological Investigations of Australian Acacia: An Ethnomedicine-Guided Bioprospective Approach; Charles Sturt University: Bathurst, Australia, 2016. [Google Scholar]
- Chatatikun, M.; Chiabchalard, A. Thai plants with high antioxidant levels, free radical scavenging activity, anti-tyrosinase and anti-collagenase activity. BMC Complement. Altern. Med. 2017, 17, 487. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Huang, C.; Yang, B.; Kallio, H.; Liu, P.; Ou, S. Regulation of phytochemicals in fruits and berries by environmental variation—Sugars and organic acids. J. Food Biochem. 2019, 43, e12642–e12660. [Google Scholar] [CrossRef]
- Ruiz-Ojeda, F.J.; Rupérez, A.I.; Gomez-Llorente, C.; Gil, A.; Aguilera, C.M. Cell models and their application for studying adipogenic differentiation in relation to obesity: A review. Int. J. Mol. Sci. 2016, 17, 1040. [Google Scholar] [CrossRef] [Green Version]
- Du, Q.; Jerz, G.; Winterhalter, P. Preparation of three flavonoids from the bark of Salix alba by high-speed countercurrent chromatographic separation. J. Liq. Chromatogr. Relat. Technol. 2004, 27, 3257–3264. [Google Scholar] [CrossRef]
- Andersen, O.M.; Markham, K.R. Flavonoids: Chemistry, Biochemistry and Applications, 1st ed.; CRC press: Boca Raton, FL, USA, 2005; pp. 10–13. [Google Scholar]
- Olsen, H.T.; Stafford, G.I.; van Staden, J.; Christensen, S.B.; Jäger, A.K. Isolation of the MAO-inhibitor naringenin from Mentha aquatica L. J. Ethnopharmacol. 2008, 117, 500–502. [Google Scholar] [CrossRef]
- Zhang, Z.; ElSohly, H.N.; Li, X.-C.; Khan, S.I.; Broedel, J.S.E.; Raulli, R.E.; Cihlar, R.L.; Walker, L.A. Flavanone glycosides from Miconia trailii. J. Nat. Prod. 2003, 66, 39–41. [Google Scholar] [CrossRef]
- Hendra, R.; Willis, A.; Keller, P.A. Phytochemical studies on the Australian native plant species Acacia pycnantha and Jacaranda mimosifolia D. Don. Nat. Prod. Res. 2019, 33, 1997–2003. [Google Scholar] [CrossRef] [Green Version]
- Imperato, F. A new chalcone glucoside and isosalipurposide from Acacia cyanophylla. Phytochemistry 1978, 17, 822–823. [Google Scholar] [CrossRef]
- Ghouila, H.; Meksi, N.; Haddar, W.; Mhenni, M.; Jannet, H. Extraction, identification and dyeing studies of Isosalipurposide, a natural chalcone dye from Acacia cyanophylla flowers on wool. Ind. Crop. Prod. 2012, 35, 31–36. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Kim, Y.S.; Choi, S.U.; Ryu, S.Y. Isolation of flavonol rhamnosides from Loranthus tanakae and cytotoxic effect of them on human tumor cell lines. Arch. Pharmacal Res. 2004, 27, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Raya-Gonzalez, D.; Pamatz-Bolanõs, T.; del Rio-Torres, R.E.; Munõz, R.E.M.; Ron-Echeverria, O.; Martinez-Pacheco, M.M. D-(+)-pinitol, a component of the heartwood of Enterolobium cyclocarpum (Jacq.). Z. Naturforsch. C J. Biosci. 2008, 63, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.B.; MacDonald, D.; Fischer, H.O. The structure of pinitol. J. Am. Chem. Soc. 1952, 74, 1479–1480. [Google Scholar] [CrossRef]
- Hudlicky, T.; Price, J.D.; Rulin, F.; Tsunoda, T. Efficient and Enantiodivergent Synthesis of (+)- and (−)-Pinitol. J. Am. Chem. Soc. 1990, 112, 9439–9440. [Google Scholar] [CrossRef]
- Chaubal, R.; Pawar, P.V.; Hebbalkar, G.D.; Tungikar, V.B.; Puranik, V.G.; Deshpande, V.H.; Deshpande, N.R. Larvicidal Activity of Acacia nilotica Extracts and Isolation of D-Pinitol–A Bioactive Carbohydrate. Chem. Biodivers. 2005, 2, 684–688. [Google Scholar] [CrossRef] [PubMed]
- Bates, S.H.; Jones, R.B.; Bailey, C.J. Insulin-like effect of pinitol. Br. J. Pharmacol. 2000, 130, 1944–1948. [Google Scholar] [CrossRef]
- Yap, A.; Nishiumi, S.; Yoshida, K.-I.; Ashida, H. Rat L6 myotubes as an in vitro model system to study GLUT4-dependent glucose uptake stimulated by inositol derivatives. Cytotechnology 2007, 55, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Lee, J.Y.; Kim, S.M.; Park, D.-A.; Jin, C.; Hong, S.P.; Lee, Y.S. A new epicatechin gallate and calpain inhibitory activity from Orostachys japonicus. Fitoterapia 2009, 80, 73–76. [Google Scholar] [CrossRef]
- Belghit, S.; Driche, E.; Bijani, C.; Zitouni, A.; Sabaou, N.; Badji, B.; Mathieu, F. Activity of 2, 4-Di-tert-butylphenol produced by a strain of Streptomyces mutabilis isolated from a Saharan soil against Candida albicans and other pathogenic fungi. J. Mycol. Med. 2016, 26, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Dharni, S.; Sanchita; Maurya, A.; Samad, A.; Srivastava, S.K.; Sharma, A.; Patra, D.D. Purification, characterisation, and in vitro activity of 2,4-di-tert-butylphenol from Pseudomonas monteilii PsF84: Conforma-tional and molecular docking studies. J. Agric. Food Chem. 2014, 62, 6138–6146. [Google Scholar] [CrossRef]
- Zhao, F.; Wang, P.; Lucardi, R.; Su, Z.; Li, S. Natural sources and bioactivities of 2,4-di-tert-butylphenol and its analogs. Toxins 2020, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Varsha, K.K.; Devendra, L.; Shilpa, G.; Priya, S.; Pandey, A.; Nampoothiri, K.M. 2,4-Di-tert-butyl phenol as the antifungal, antioxidant bioactive purified from a newly isolated Lactococcus sp. Int. J. Food Microbiol. 2015, 211, 44–50. [Google Scholar] [CrossRef]
- Khiralla, A.; Spina, R.; Varbanov, M.; Philippot, S.; Lemiere, P.; Slezack-Deschaumes, S.; André, P.; Mohamed, I.; Yagi, S.M.; Laurain-Mattar, D. Evaluation of antiviral, antibacterial and antiproliferative activities of the endophytic fungus Curvularia papendorfii, and isolation of a new polyhydroxyacid. Microorganisms 2020, 8, 1353. [Google Scholar] [CrossRef]
- Chuah, T.; Norhafizah, M.; Ismail, B. Evaluation of the biochemical and physiological activity of the natural compound, 2,4-ditert-butylphenol on weeds. Crop Pasture Sci. 2015, 66, 214–223. [Google Scholar] [CrossRef]
- Huang, X.; Chao, L.; Jiang, P.; Zhang, X.; Zhang, X.; Qin, P.; Tian, W. Identification of chrysanthemum root exudates and allelopathic effects of the three plants. Hubei Agric. Sci. 2017, 56, 1061–1071. [Google Scholar]
- Abd El Gawad, Y.; El-Amier, A. Allelopathy and potential impact of invasive Acacia saligna (Labill.) wendl. on plant diversity in the Nile delta coast of Egypt. Int. J. Environ. Res. 2015, 9, 923–932. [Google Scholar]
- Kamel, M.; Hammad, S.A. The Allelopathic Effect of the Exotic Tree Acacia saligna on the Germination of Wheat and Canola. Ecol. Balk. 2015, 7, 103–112. [Google Scholar]
- Hwang, I.W.; Chung, S.K. Isolation and identification of myricitrin, an antioxidant flavonoid, from daebong persimmon peel. Prev. Nutr. Food Sci. 2018, 23, 341. [Google Scholar] [CrossRef]
- Cai, R.; Li, X.; Chen, B.; Xie, Y.; Xie, H.; Chen, D. Antioxidant change in biosynthesis from naringenin chalcone to flavonoid apingenin. Chem. Sel. 2019, 4, 5155–5159. [Google Scholar] [CrossRef]
- Ouyang, X.; Li, X.; Lu, W.; Zhao, X.; Chen, D. A null B-ring improves the antioxidant levels of Flavonol: A comparative study between Galangin and 3, 5, 7-Trihydroxychromone. Molecules 2018, 23, 3083. [Google Scholar] [CrossRef] [Green Version]
- Domitrović, R.; Rashed, K.; Cvijanović, O.; Vladimir-Knežević, S.; Škoda, M.; Višnić, A. Myricitrin exhibits antioxidant, anti-inflammatory and antifibrotic activity in carbon tetrachloride-intoxicated mice. Chem.-Biol. Interact 2015, 239, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhu, X.; Lu, Q.; Zhang, L.; Wang, X.; Liu, R. C-ring cleavage metabolites of catechin and epicatechin enhanced antioxidant activities through intestinal microbiota. Food Res. Int. 2020, 135, 109271–109281. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Li, X.; Zhao, C.; Gao, X.; Wang, Y.; Gao, W. Active compounds, antioxidant activity and α-glucosidase inhibitory activity of different varieties of Chaenomeles fruits. Food Chem. 2018, 248, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Jiang, Q.; Wang, T.; Liu, J.; Chen, D. Comparison of the antioxidant effects of quercitrin and isoquercitrin: Understanding the role of the 6″-OH group. Molecules 2016, 21, 1246. [Google Scholar] [CrossRef]
- Hong, C.-O.; Lee, H.A.; Rhee, C.H.; Choung, S.-Y.; Lee, K.-W. Separation of the antioxidant compound quercitrin from Lindera obtusiloba Blume and its antimelanogenic effect on B16F10 melanoma cells. Biosci. Biotechnol. Biochem. 2013, 77, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Priscilla, D.; Roy, D.; Suresh, A.; Kumar, V.; Thirumurugan, K. Naringenin inhibits a-glucosidase activity: A promising strategy for the regulation of postprandial hy-perglycemia in high fat diet fed streptozotocin induced diabetic rats. Chem.-Biol. Interact. 2014, 210, 77–85. [Google Scholar] [CrossRef]
- Zhang, L.-L.; Han, L.; Yang, S.-Y.; Meng, X.-M.; Ma, W.-F.; Wang, M. The mechanism of interactions between flavan-3-ols against a-glucosidase and their in vivo antihyperglycemic effects. Bioorg. Chem. 2019, 85, 364–372. [Google Scholar] [CrossRef]
- Gong, T.; Yang, X.; Bai, F.; Li, D.; Zhao, T.; Zhang, J.; Sun, L.; Guo, Y. Young apple polyphenols as natural α-glucosidase inhibitors: In vitro and in silico studies. Bioorg. Chem. 2020, 96, 103625–103635. [Google Scholar] [CrossRef]
- Tadera, K.; Minami, Y.; Takamatsu, K.; Matsuoka, T. Inhibition of alpha-glucosidase and alpha-amylase by flavonoids. J. Nutr. Sci. Vitaminol. 2006, 52, 149–153. [Google Scholar] [CrossRef]
- Proença, C.; Freitas, M.; Ribeiro, D.; Oliveira, E.; Sousa, J.; Tomé, S.; Ramos, M.; Silva, A.; Fernandes, P.; Fernandes, E. α-Glucosidase inhibition by flavonoids: An in vitro and in silico structure–activity relationship study. J. Enzym. Inhib. Med. Chem. 2017, 32, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Liu, X.; Jiang, Z.; Geng, S.; Ma, H. Interaction Mechanism of Flavonoids and α-Glucosidase: Experimental and Molecular Modelling Studies. Foods 2019, 8, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Hidalgo, M.; León-González, A.J.; Gálvez-Peralta, M.; González-Mauraza, N.H.; Martin-Cordero, C. D-Pinitol: A cyclitol with versatile biological and pharmacological activities. Phytochem. Rev. 2021, 20, 211–224. [Google Scholar] [CrossRef]
- Matsui, T.; Tanaka, T.; Tamura, S.; Toshima, A.; Tamaya, K.; Miyata, Y.; Tanaka, K.; Matsumoto, K. α-Glucosidase inhibitory profile of catechins and theaflavins. J. Agric. Food Chem. 2007, 55, 99–105. [Google Scholar] [CrossRef]
- Giorgio, E.; Parrinello, N.; Caccamese, S.; Rosini, C. Non-empirical assignment of the absolute configuration of (−)-naringenin, by coupling the exciton analysis of the circular dichroism spectrum and the ab initio calculation of the optical rotatory power. Org. Biomol. Chem. 2004, 2, 3602–3607. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, D.; Ma, X.; Jiang, F.; He, Q.; Qiu, S.; Li, Y.; Wang, G. Ionic liquid–ultrasound-based extraction of biflavonoids from Selaginella helvetica and investigation of their antioxidant activity. Molecules 2018, 23, 3284. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.G.; Bishop, K.S.; Tanambell, H.; Buchanan, P.; Smith, C.; Quek, S.Y. Characterisation of the bioactivities of an ethanol extract and some of its constituents from the New Zealand native mushroom Hericium novae-zealandiae. Food Funct. 2019, 10, 6633–6643. [Google Scholar] [CrossRef]
- Konan, K.; Le Tien, C.; Mateescu, M. Electrolysis-induced fast activation of the ABTS reagent for an antioxidant capacity assay. Anal. Methods 2016, 8, 5638–5644. [Google Scholar] [CrossRef]
- Re, R.; Pellegrini, N.; Proteggente, A.; Pannala, A.; Yang, M.; Rice-Evans, C. Antioxidant activity applying an improved ABTS radical cation decolorization assay. Free Radic. Biol. Med. 1999, 26, 1231–1237. [Google Scholar] [CrossRef]
- Ning, Z.-W.; Zhai, L.-X.; Huang, T.; Peng, J.; Hu, D.; Xiao, H.-T.; Wen, B.; Lin, C.-Y.; Zhao, L.; Bian, Z.-X. Identification of α-glucosidase inhibitors from cyclocarya paliurus tea leaves using UF-UPLC-Q/TOF-MS/MS and molecular docking. Food Funct. 2019, 10, 1893–1902. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Extract | Mass of Extract (g), Colour | ||
|---|---|---|---|
| Dried Flowers (FL) | Dried Leaves (LF) | Dried Barks (BK) | |
| Hexane (hex) | 1.71, yellow | 3.08, dark green | 0.68, yellow |
| Dichloromethane (DCM) | 1.79, green | 4.98, green | 2.12, green |
| Methanol (MeOH) | 26.16, yellow | 25.37, green-yellow | 18.26, yellow |
| Water (H2O) | 36.31, yellow | 13.32, yellow | 4.34, yellow |
| Extracts | IC50 (µg/mL) | |
|---|---|---|
| DPPH Assay | ABTS●+ Assay | |
| FL-MeOH | 331.5 ± 17.21 **** | 316.6 ± 11.45 **** |
| LF-MeOH | 190.1 ± 59.15 **** | 146.7 ± 0.99 |
| BK-MeOH | 94.24 ± 19.89 | 55.44 ± 6.84 |
| Vitamin C | 49.97 ± 10.76 | 72.25 ± 4.42 |
| Extracts | IC50 (µg/mL) |
|---|---|
| FL-MeOH | 34.93 ± 2.67 *** |
| LF-hex | 285.5 ± 100.9 |
| LF-MeOH | 38.69 ± 1.01 *** |
| BK-hex | 289.9 ± 29.17 |
| BK-MeOH | 4.37 ± 0.24 **** |
| BK-H2O | 23.27 ± 3.88 **** |
| Acarbose | 254 ± 22.18 |
| ID | 1H (δ ppm, m, J in Hz, Integration) | δ 13C (ppm) | COSY | TOCSY | NOESY | HMBC(C→H) a |
|---|---|---|---|---|---|---|
| 2 | 5.35 (td; 12.96, 2.8; 1H) | 80.48 | H3a H3b | H3a, H3b | H3b (strong), H3a (weak) | C4, C2′, C6′ |
| 3a | 2.70 (td; 17.64, 2.95; 1H) | 46.45 | H2, H3b | H2, H3b | H2 | C4, C10 |
| 3b | 3.04 (m; 1H) | H2, H3a | H2, H3a | H2 | C4, C2, C1′ | |
| 4 | 193.22 | |||||
| 5 | 166.70 | |||||
| 6 | 6.15 (d; 2.24; 1H) | 99.56 | H6 | H8 | H8 | C4, C5, C8 C10 |
| 7 | 167.16 | |||||
| 8 | 6.50 (d; 2.2; 1H) | 100.49 | H8 | H6 | H6 | C4, C7, C6, C9, C10 |
| 9 | 162.51 | |||||
| 10 | 107.21 | |||||
| 1′ | 131.12 | |||||
| 2′ | 7.31 (dd; 8.56, 3.16; 1H) | 129.14 | H3′, H6′ | H3′, H6′ | H3′ (strong), H2 (weak) | C2, C1′, C3′, C4′ |
| 3′ | 6.83 (dd; 8.64, 1.96; 1H) | 116.46 | H2′, H5′ | H2′, H5′ | H2′ | C2′, C4′ |
| 4′ | 159.13 | |||||
| 5′ | 6.83 (dd; 8.64, 1.96; 1H) | 116.46 | H3′, H6′ | H3′, H6′ | H6′ | C6′, C4′ |
| 6′ | 7.31 (dd; 8.56, 3.16; 1H) | 129.14 | H2′, H5′ | H2′, H5′ | H5′ | C2, C1′, C5′, C4′ |
| 1” | 4.78 (d; 7.48; 1H) | 105.16 | H2″ | H2”, H3”, H4” | H8 (strong), H2″, H3″, H4″ | C7, C2″ |
| 2” | 3.57 (m; 1H) | 74.81 | H1″ | H1″, H3″, H4″, H5″a, H5b″ | H1″, H5″a | |
| 3” | 3.45 (t; 8.4; 1H) | 78.79 | H1″, H2″, H4″, H5″a, H5b″ | H1″, H5″a, H5″b | C2″ | |
| 4” | 3.51 (t; 8.08; 1H) | 77.3 | H5″b | H1″, H2″, H3″, H5″a, H5b″ | H1″, H5″a, H5″b | C2″ |
| 5”a | 3.97 (m; 1H) | 62.68 | H5″b | H2″, H3″, H4″, H5b″ | H2″, H3″, H4″, H5″b | |
| 5”b | 3.78 (m; 1H) | H4″, H5″a | H2″, H3″, H4″, H5a″ | H3″, H4″, H5″a |
| ID | 1H (δ ppm, m, J Hz, Integration) | 13C (δ ppm) | COSY | TOCSY | HMBC(C→H) a | NOESY |
|---|---|---|---|---|---|---|
| 2 | 175.27 | |||||
| 3 | 3.86 (dd; 11.8, 3.68; 1H) | 53.90 | H4b, H4b | H5, H4a, H4b | C2, C4, C5 | H4a (strong), H4b |
| 4a | 2.16 (m; 1H) | 32.74 | H3, H4b, H5 | H3, H5, H4b | H3 (strong), H4b (strong) | |
| 4b | 1.91 (m; 1H) | H3, H4a, H5 | H3, H5, H4a | C2, C3, C5, C6 | H5 (strong), H4a | |
| 5 | 4.18 (m; 1H) | 61.80 | H4a, H4b, H6 | H3, H4a, H4b, H6, H7 | H4b (strong), H6 (strong) | |
| 6 | 1.85 (m; 2H) | 26.50 | H5, H7 | H5, H7 | C4, C5, C7 | H5 |
| 7 | 3.26 (m; 2H) | 38.54 | H6 | H5, H6 | C5, C6 | H6 (strong) |
| Compounds | IC50 against DPPH (µM) | IC50 against ABTS (µM) |
|---|---|---|
| Isosalipurposide 3 | 1559 ± 28.16 *** | 1686 ± 95.26 **** |
| Naringenin 1 | >10,000 a | 1525 ± 316.50 **** |
| Quercitrin 4 | 322.6 ± 14.05 **** | 355.3 ± 12.08 |
| Myricitrin 8 | 199.9 ± 4.83 **** | 285.9 ± 7.21 |
| Naringenin-7-O-α-L-arabinopyranose 2 | >10,000 a | 4146 ± 99.15 **** |
| D-(+)-pinitol 5a | 1675 ± 65.72 **** | 475 ± 24.20 |
| (−)-Pinitol 5b | 6865 ± 69.08 **** | 2096 ± 70.40 **** |
| (−)-Epicatechin 6 | 278 ± 8.62 **** | 92.58 ± 13.03 |
| 2,4-Di-t-butylphenol 7 | >10,000 a | 2715 ± 64.02 **** |
| 3-hydroxy-5-(2-aminoethyl)-dihydrofuran-2(3H)-one 9 | >10,000 a | >10,000 a |
| Vitamin C | 1072 ± 47.64 | 460.2 ± 56.29 |
| Compound | IC50 (μM) |
|---|---|
| Isosalipurposide 3 | 116.5 ± 26.40 |
| Naringenin 2 | 89.71 ± 10.22 * |
| Quercitrin 4 | 177.3 ± 11.34 |
| Myricitrin 8 | 351.6 ± 24.88 |
| Naringenin-7-O-α-L-arabinopyranose 2 | 769.1 ± 95.82 **** |
| D-(+)-pinitol 5a | 74.69 ± 0.23 * |
| (−)-pinitol 5b | 164.2 ± 8.36 |
| (−)-Epicatechin 6 | 63.58 ± 11.83 * |
| 2,4-Di-t-butylphenol 7 | 259 ± 58.34 |
| 3-hydroxy-5-(2-aminoethyl) dihydrofuran-2(3H)-one 9 | >1000 a |
| Acarbose | 239.9 ± 31.74 |
| Cell growth | 100% confluent | Lipid droplets formation and development | Excessive lipid droplets | |||
| 48 h | 48 h (day −2 to 0) | 48 h (day 0 to 2) | 48 h (day 2 to 4) | 48 h (day 4 to 6) | 48h (day 6 to 8) | Between day 8 & 12 |
| Feed M1 | Feed M1 | Feed M2 | Feed M3 | Feed M3 | Feed M4 | Treatment with extracts/compounds |
Preadipocyte mature adipocyte mature adipocyte | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asmara, A.P.; Prasansuklab, A.; Tencomnao, T.; Ung, A.T. Identification of Phytochemicals in Bioactive Extracts of Acacia saligna Growing in Australia. Molecules 2023, 28, 1028. https://doi.org/10.3390/molecules28031028
Asmara AP, Prasansuklab A, Tencomnao T, Ung AT. Identification of Phytochemicals in Bioactive Extracts of Acacia saligna Growing in Australia. Molecules. 2023; 28(3):1028. https://doi.org/10.3390/molecules28031028
Chicago/Turabian StyleAsmara, Anjar P., Anchalee Prasansuklab, Tewin Tencomnao, and Alison T. Ung. 2023. "Identification of Phytochemicals in Bioactive Extracts of Acacia saligna Growing in Australia" Molecules 28, no. 3: 1028. https://doi.org/10.3390/molecules28031028