
On the Shoulders of Giants—Reaching for Nitrogenase

Institute of Biochemistry, Albert-Ludwigs-Universität Freiburg, Albertstrasse 21, 79104 Freiburg im Breisgau, Germany
Molecules 2023, 28(24), 7959; https://doi.org/10.3390/molecules28247959
Submission received: 8 October 2023
/
Revised: 14 November 2023
/
Accepted: 27 November 2023
/
Published: 5 December 2023
(This article belongs to the Special Issue Molybdenum and Tungsten Enzymes—State of the Art in Research)
Abstract
:Only a single enzyme system—nitrogenase—carries out the conversion of atmospheric N2 into bioavailable ammonium, an essential prerequisite for all organismic life. The reduction of this inert substrate at ambient conditions poses unique catalytic challenges that strain our mechanistic understanding even after decades of intense research. Structural biology has added its part to this greater tapestry, and in this review, I provide a personal (and highly biased) summary of the parts of the story to which I had the privilege to contribute. It focuses on the crystallographic analysis of the three isoforms of nitrogenases at high resolution and the binding of ligands and inhibitors to the active-site cofactors of the enzyme. In conjunction with the wealth of available biochemical, biophysical, and spectroscopic data on the protein, this has led us to a mechanistic hypothesis based on an elementary mechanism of repetitive hydride formation and insertion.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction: You Cannot Fix Nitrogen with an Enzyme
The reductive fixation of atmospheric nitrogen by a single known enzyme, nitrogenase, is a story of many heroes, sung and unsung, and a roller-coaster ride of discoveries, disappointments, frustrations, and break-throughs that started as far back as 1888, when Hellriegel and Willfarth reported nitrogen fixation by legumes and graminees [1]. This turned out to be a bacterial process, and in 1903, Jacob Lipman described a free-living soil bacterium he found near Vineland, New Jersey, to be a ‘diazotroph’, a N2-eater [2]. This organism, Azotobacter vinelandii, remains the most extensively studied model for biological nitrogen fixation to this day, as it is easily cultivated as an obligate aerobe that produces high amounts of nitrogenase during N-limited growth.
In more than one way, the term ‘biological nitrogen fixation’ is an oxymoron, and nitrogenase, the only known enzyme that catalyzes it, should not be able to do so. Life is a play that is enacted within a rather small window of biophysical boundaries and is based on a few fundamental principles that are indisputable. All organisms are energy-converting machines that take up one form of energy from their environment and convert it into something useful, most prominently the phosphodiester bonds of adenosine-5′-triphosphate (ATP), our basic currency of metabolic energy. In essence, however, we are electrochemical cells that gain energy from the oxidation of nutrients and store it by reducing oxidized compounds. The actual energetic small change that we are shuffling around is electrons that are transferred between redox couples of different midpoint potentials, back and forth, as long as we live, as doing this is the actual definition of being alive. Life at some stage then made the—questionable—decision to leave the oceans, but even so, our cells have remained complex chemical reactors that operate in an aqueous, mildly saline milieu, around neutral pH, and within boundaries of temperature and pressure that allow for water to stay in the liquid phase—‘ambient conditions’, as we usually call it. The protic solvent water, however, puts strict limitations on our energy metabolism. The splitting of water according to
requires an overpotential of at least 1.23 V (vs. the standard hydrogen electrode, SHE) at pH 0, but only about 0.82 V vs. SHE at a physiological pH of 7. Nowadays, this reaction is widely discussed as a means of storing solar energy as fuel H2, but its biochemical relevance is that in an aqueous milieu, cells should not be able to reduce any substrate that requires a more negative potential than 0.82 V, corresponding to just below 80 kJ·mol–1 for a single-electron reduction. Electrons with a more negative potential much rather reduce water, which is always present in high excess. Evolution over time has splendidly exploited this ‘water window’, optimally so in the aerobic respiration of our mitochondria, which is the most efficient biological energy metabolism we know. However, there are some essential ingredients to the soup of life that cannot be accessed under these conditions, and the most prominent of these finally gets us back on track: The element nitrogen is a key requirement for building biological macromolecules such as nucleic acids and proteins, but almost all (>99%) of the nitrogen in Earth’s biosphere is present in the form of the chemically inert dinitrogen gas, N2, that makes up 78% of our atmosphere [3]. Triple-bonded N2 has a bond dissociation energy of −946 kJ mol−1, which implies that its reduction requires an overpotential of at least 1.63 V. Reduction of nitrogen at ambient conditions in an aqueous milieu should not be possible, and nitrogenase, which does just that is an impossible enzyme. Enzymes surely have their little tricks and cheats to get things going, but at this magnitude, thermodynamics are not negotiable. The problem is only emphasized by the prominent Haber–Bosch process of industrial nitrogen fixation, invented in 1906 and first established at BASF in 1913 [4], which takes LeChatelier’s principle to the task and reacts N2 and H2 gas at an iron catalyst surface at temperatures of 400 to 650 °C and 200–400 atm pressure.
2 H2O → 2 H2 + O2
N2 + 3 H2 → 2 NH3
This surely works famously well, but it is no option for a microorganism in need of bioavailable ammonium cations.
2. Have You Tried to Crystallize the Protein?
Biological nitrogen fixation was one of my smaller worries in 1992 as a student of Biology at the University of Konstanz in the very south of what had just recently ceased to be ‘West’ Germany. I recall a day in a lab course on plant biochemistry run by the team of Peter Böger [5] when there was a buzz of excitement among the graduate students who served as teaching assistants. The experiment of the day was about biological nitrogen fixation, monitoring oxidative stress on the ammonia production by a diazotrophic cyanobacterium that was catalyzed by nitrogenase, and obviously, someone in the United States had produced a crystal structure of this enzyme from Azotobacter vinelandii that defied all expectations, revealing am unprecedented catalytic center that must somehow be key to the unique abilities of this system [6,7]. At the time, enzymology and mechanism were on everyone’s agenda in Konstanz, and so I became part of the first generation of students who had to learn to draw this Escher-like two-dimensional projection of the FeMo cofactor of nitrogenase on paper for my exam.
The structure of nitrogenase was ground-breaking indeed, and the one who had achieved this was, of course, Doug Rees at Caltech. In short succession, the Rees group reported the architecture of the nitrogenase clusters [6], the three-dimensional structure of MoFe protein [7], and the one for Fe protein [8], the second component of the nitrogenase system. Crystal structures were a luxury at the time, the product of hard work both in the laboratory and on the computer, and the information they could provide was truly invaluable. Nitrogenase was first isolated in 1966 by Bulen and LeComte [9], and a wealth of physiological and biochemical information has been accumulated since. Structural Biology now brought many of these findings together. The nitrogenase system consists of two component proteins; the dinitrogenase reductase binds and hydrolyzes ATP and provides low-potential electrons by forming a transient complex with the second component, dinitrogenase (Figure 1). Three isoforms of nitrogenases are known, utilizing Mo, V, or Fe as the apical ion to their active-site cofactor, and under conditions of low electron flux and high pN2, all catalyze the reduction of dinitrogen according to
N2 + 10 H+ + 8 e− + 16 ATP → 2 NH4+ + H2 + 16 [ADP + Pi].
In this reaction, electron transfer occurs sequentially, and the stoichiometric by-product H2 is of critical mechanistic relevance, as discussed below. The Rees structures had clarified the architecture of the metal cofactors, finding that both the electron-transferring P-cluster and the catalytic FeMo cofactor are unique to this enzyme, and the general expectation at the time was that this information would advance understanding of biological nitrogen fixation to a similar degree as the structure of the photosynthetic reaction center of Rhodopseudomonas viridis had for photosynthesis a few years prior [10].
In my studies in Konstanz, I moved on from this first encounter with nitrogenase, but not too far. During my last year, I worked as a student assistant in the group of Peter Kroneck, together with his graduate student Frank Neese, whose day job was to study the copper enzyme nitrous oxide reductase while teaching himself quantum chemistry in his spare time to move on to become one of the world leaders in this field [11]. During this time, I heard a lecture by Albrecht Messerschmidt from Martinsried, who had collaborated with Peter to investigate ascorbate oxidase, one of the first multicopper oxidases characterized at this level [12,13]. I was fascinated with protein crystallography as a method, and Peter and Albrecht soon offered me the opportunity to perform my diploma thesis work in Martinsried, in the Department of Structural Research headed by Robert Huber, who had been awarded the 1988 Noble Prize in Chemistry for the structure of the aforementioned photosynthetic reaction center, the first membrane protein crystal structure ever reported [10]. This was the year 1996, and crystallography’s triumphant sweep through the molecular life sciences was just picking up speed. A year prior, Juan Fontecilla-Camps and co-workers solved the first structure of Ni,Fe-hydrogenase [14], and Doug Rees had reported the first tungsten-containing enzyme, AOR [15], followed shortly thereafter by the first molybdoenzyme from Robert’s group [16]. My own task that I carried over into my Ph.D. work with Robert and Peter was the pentaheme cytochrome c nitrite reductase, an ammonium-producing enzyme from the energy-conserving pathway of dissimilatory nitrite reduction [17]. At this time, metalloproteins were almost exclusively isolated from their native source, chromatography techniques were less automated, synchrotron radiation sources were far weaker and less reliable than they are now, and computers… well, computers. It was the 1990s. Evans and Sutherland vector graphics machines and VAXstations slowly gave way to incredibly powerful SGI workstations that could refine a small structure in only a day or so. Instead of smartphones, the digital time killer of the day was the Tamagotchi (Google it!). There was no world-wide web, and the Internet was a fledgling, text-based labyrinth for only the nerdiest among us. In our Bavarian lab, every submitted compute job would provide ample time to visit a beer garden with good conscience, and a new structure was the product of an entire doctoral project. Metalloproteins offered the possibility to combine structural and biochemical information with advanced spectroscopic methods to provide a comprehensive picture of a complex metabolic machine. So, after obtaining my Ph.D. in 2000, I knew that my next challenge should be again in the field of metalloprotein structural biology. This was when I remembered nitrogenase. Doug Rees was a household name in Martinsried: He once almost joined Robert’s group as a postdoc to analyze crystals of a certain reaction center but then decided to work with Jim Howard on nitrogen fixation. I contacted Doug Rees at Caltech, went to an interview, and gladly accepted the position he offered me.
3. A Closer Look
With an entire laboratory dedicated to the isolation and analysis of nitrogenase, the Rees group had a well-operated setup that allowed me to jump right into my work. Together with Susana Andrade, Benedikt Schmid, and Akif Tezcan, and supported by technician Mika Walton, we isolated Mo-nitrogenase and studied aspects of its assembly, complex formation, and function. Crystals at the time were still grown in sealed capillaries within an anoxic glove box, from which they had to be harvested with gloved hands, cryoprotected, and frozen in liquid nitrogen (pure substrate!). This was tedious but not uncommon at the time, and the oxygen sensitivity of nitrogenase was legendary. Soon, however, I decided to put that particular legend to the test. Having worked with O2-sensitive N2O reductase in Konstanz and Martinsried, we set up regular sitting drop vapor diffusion plates in a glove box. Over lunch, multiple MoFe protein crystals had formed, and it was unproblematic to optimize these into large single crystals of 1–2 mm in length. These crystals diffracted to almost 1.1 Å resolution at the Stanford synchrotron source, but initially, we hardly expected more than a cosmetic improvement over the then-available 2.0 Å structure of the enzyme [18]. To our surprise, the high-quality electron density map revealed an additional maximum in the center of the cofactor that we soon understood to be a µ6-coordinated light atom at the heart of the cluster that was obfuscated at lower resolution by Fourier series termination artifacts created by the highly symmetric cluster itself [19]. This re-defined our picture of the cofactor architecture (Figure 2), but in terms of understanding nitrogenase catalysis, if anything, it made matters worse, as the light atom occupied the only remaining open coordination site to the metal ions of the cluster. Even at atomic resolution, nitrogenase remained reluctant to reveal its inner secrets. Another outstanding feature of the Rees lab was Jim Howard. Having retired from the University of Minnesota, Doug’s former postdoc advisor understandably spent the winter months in Southern California, and during this time, Jim was fully involved in the ongoing work in nitrogenase. As a true enzymologist, he was a constant source of ideas and a (very) rigorous experimenter and running activity assays with Jim was a great experience. Jim passed away in 2022 and is dearly missed.
With the brand-new high-resolution data, Doug Rees sent us to the 2002 GRC on Nitrogen Fixation, which turned out to be the last of its name. It was chaired by Brian Hales, who generously gave me an extra speaker slot on Monday morning. On the opening night on Sunday, I learned much about the restrictions for alcoholic beverages on college campuses and received a stern warning from Barry Smith that I would have difficulties convincing the crowd of my findings. Fortunately, there is little ambiguity in atomic-resolution crystal structures, and the news was taken up very well by the crowd. Barry later wrote a wonderful perspective for our paper [20]. At the meeting, I emphasized the result of our analysis that this central atom is most likely nitrogen more forcefully than we did in the publication, and this, of course, later turned out to be incorrect—the electron density anomaly that hid the central atom at resolutions below 1.5 Å (Figure 2b) would also enhance it slightly at 1.16 Å—but the presence of a central light atom did much to explain the unusual stability of the cofactor that can be extracted in an intact state from the enzyme and was very well received in particular by synthetic and theoretical chemists (Figure 2c). The other part of the experience of this first GRC I attended was to meet many of the literal giants on whose shoulders we stood, starting to make our own contributions. There were David Lowe and Roger Thorneley, Barry Smith and Bob Eady, Bill Newton and Dennis Dean, Dimitri Coucouvanis, Vince Huynh, Lance Seefeldt, Brian Hoffman, Paul Ludden, Stephen Cramer, Dick Cammack, Mike Johnson, Juan Fontecilla, and many more. And, among them, many juniors proved that although the meeting changed its name (to iron–sulfur enzymes and later to metallocofactors), the torch of nitrogenase research was carried on. Patricia dos Santos, Markus Ribbe, Luis Rubio, John Peters, Cathy Drennan, Pat Holland, and Akif Tezcan were early in their careers and have since done so much to explore new avenues of nitrogenase mechanism and application. They are giants standing on the shoulders of giants, which is not necessarily the most stable situation, but it sure is motivating to look around you and make sure that you will not only see knees. The work of the participants of this meeting and of many others has provided an incredible wealth of high-quality data, and part of what makes the current state of the field today so exciting is that, in many cases, we can now reap the harvest of their efforts and connect the dots to obtain a comprehensive picture of what happens during catalysis in nitrogenase.
Figure 2.
Evolution of structural models for the FeMo cofactor of Mo-nitrogenase. (a) The original model by Kim and Rees showed the cofactor with one µ2-bridging ligand unassigned [6]. (b) Improving the structure to 2.0 Å resolution, the bridging ligand was identified as a third bridging sulfide, S5A [18]. (c) Only with an atomic-resolution structure, the Fourier series termination artifacts caused by the high-symmetry structure of the cluster were overcome and a central light atom was revealed [19]. (d) Through a combination of HERFD-XAS, ESEEM, and high-resolution crystallography, the central light atom was identified as a carbide in 2011 [21,22].
Figure 2.
Evolution of structural models for the FeMo cofactor of Mo-nitrogenase. (a) The original model by Kim and Rees showed the cofactor with one µ2-bridging ligand unassigned [6]. (b) Improving the structure to 2.0 Å resolution, the bridging ligand was identified as a third bridging sulfide, S5A [18]. (c) Only with an atomic-resolution structure, the Fourier series termination artifacts caused by the high-symmetry structure of the cluster were overcome and a central light atom was revealed [19]. (d) Through a combination of HERFD-XAS, ESEEM, and high-resolution crystallography, the central light atom was identified as a carbide in 2011 [21,22].

From Pasadena, I accepted a position as a junior (assistant) professor at the University of Göttingen, Germany, in 2003. The junior professorships were a new career path in the German academic system that no one really had a clear concept of, me least of all. I was, however, in the lucky position to work in the newly established Department of Molecular Structural Biology, headed by Ralf Ficner, who provided all the support and resources that a starting group leader could ask for. With my own lab, our focus shifted away from nitrogenase for the time being as we studied various membrane proteins and transporters involved in the nitrogen cycle. I kept following the nitrogenase field closely but did not contribute. During this time, Tezcan and Rees published their excellent work on the complex formation between different nitrogenase components [23], Ribbe and Hu [24,25,26], as well as Rubio [27,28] studied the assembly of the nitrogenase components, and Seefeldt, Hoffmann and Dean drew the first outlines of their proposal of hydride accumulation on the cofactor that would be a cornerstone of our current mechanistic understanding [29]. Many other important findings were made in this flourishing field during this time, and when I accepted an offer for a full professorship as chair in Biochemistry at the University of Freiburg in 2008, returning to nitrogenase was high up on my agenda.
4. Episode IV: A New Beginning
Working once more in a chemistry department, we built up cell growth, anoxic protein biochemistry, and protein crystallography, and most importantly, I was able to attract many talented, bright, and highly dedicated students to work on the various aspects of structural bioinorganic chemistry that we wanted to address. The first question was about the actual nature of the central light atom I had found in the Rees lab. Brian Hoffman’s group has since provided data to show that our initial interpretation as a nitrogen species was not correct [30,31], and three years down the road, almost a decade after its discovery, Kyle Lancaster and Serena DeBeer at Cornell studied MoFe protein by HERFD-XAS, while we kept working on atomic-resolution X-ray crystallography and ESEEM spectroscopy in collaboration with Stefan Weber. We frequently discussed our data and realized that it all pointed at the central atom being carbon. Teaming up, these results were then condensed into a back-to-back study [21,22]. Shortly thereafter, Markus Ribbe and Yilin Hu, who had provided protein for Serena’s work, showed that this interstitial carbide originated from S-adenosyl methionine and was inserted by the radical/SAM enzyme NifB, another unique reaction in the nitrogenase world [32]. As exciting as these findings were for the experimentalists, they underlined once more that most work so far had only addressed the resting state of Mo-nitrogenase, where there is no free coordination site on any of the metal ions. The mode and position of nitrogen binding were still up for grabs, and the mechanism of biological nitrogen fixation remained as enigmatic as ever. Twenty years after the first crystal structure of Mo-nitrogenase, the gold standard framework for nitrogenase catalysis still was the epochal enzymological analysis by David Lowe and Roger Thorneley that led to a kinetic scheme for the enzyme with eight intermediate states representing the individual electron transfer steps [33,34,35,36] (Figure 3). Structural biology had contributed much to our understanding of the resting state, E0, but little beyond.
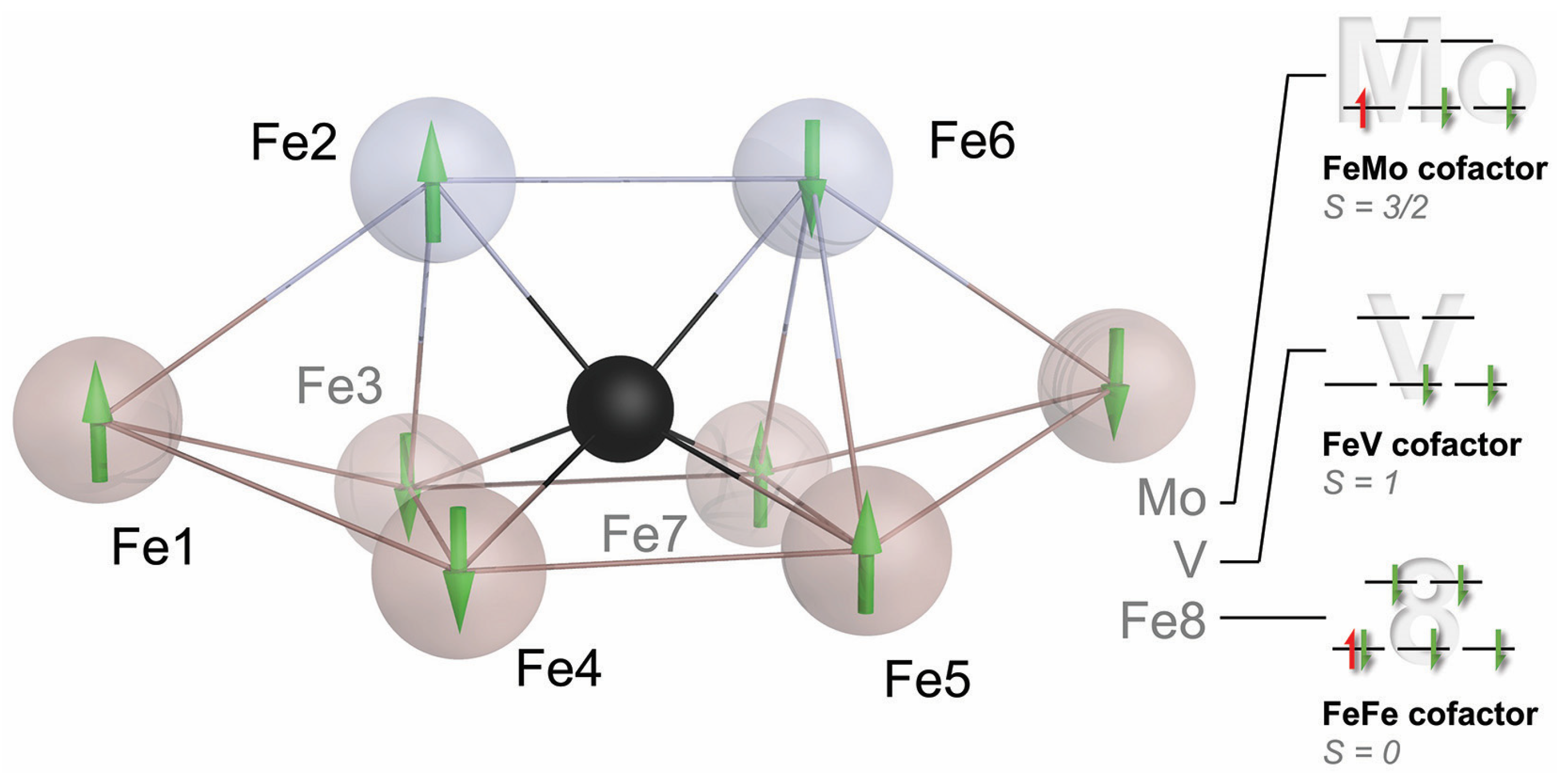
The new and complete structure of the cofactor also triggered multiple theory groups to study its electronic structure and properties, but the difficulties in treating such complex systems led to quite divergent mechanistic proposals. The road forward was not immediately apparent. In its resting state, nitrogenase does not bind substrates, and according to Lowe and Thorneley (Figure 3), it will not even bind N2 prior to reaching E4, a high-energy state that will be extremely hard to access by structural analysis. We, therefore, proceeded to further characterize the electronic structure of the FeMo cofactor to provide a better basis for theory and to help us better understand what makes this metal site so unique. It was time to pick up on some unfinished business from the Caltech days. The first of these was an EPR study using single crystals of MoFe protein, aiming to derive a spatial correlation between the pseudo-D3 symmetric structure of the FeMo cofactor and the apparent S = 3/2 g-tensor of its resting state E0. Susana Andrade and Thomas Spatzal collected EPR and diffraction data for Mo-nitrogenase, showing that the rhombic g-tensor indeed aligned specifically such that its longest axis was collinear with the pseudo-three-fold of the cofactor. We noted that the other two principal axes of the apparent g-tensor that were very different with gy = 3.65 and gx = 2.01 were oriented to highlight two irons of the cofactor, Fe2 and Fe6, in the direction of gx, while gy was in a plane formed by four others, Fe3, Fe4, Fe5, and Fe7 [38]. Like many findings on nitrogenase, this first indication of a special role for Fe2 and Fe6 would take years more to gain significance. With Doug Rees, I expanded on an idea that Holger Dobbek had conceived during our time as graduate students in Martinsried: As the anomalous scattering contribution Δf″ of an atom of a given element is proportional to its absorption cross-section for X-rays, it should change among diffraction datasets collected along an X-ray absorption edge for this element. With the added spatial resolution of a diffraction experiment, this allows for the reconstruction of an individual X-ray absorption curve for each scattering atom of a given type. While such data are not as highly resolved as X-ray absorption spectroscopy, our proof-of-principle study on a [2Fe:2Fe] ferredoxin showed that differences such as oxidation states can be very well distinguished [39]. In application to nitrogenase, Thomas Spatzal collected a series of diffraction datasets along the K-edge of iron and refined the individual Δf″ contributions for each iron site at every wavelength [40]. At the same time, Ragnar Björnsson and Serena DeBeer, now a director at the Max Planck Institute for Chemical Energy Conversion, reinvestigated the single molybdenum ion in the cluster by Mo-K-edge XAS. Her data provided strong support for a Mo(III) ion in the cluster, more reduced than any other known Mo site in biology, where the metal typically oscillates between Mo(IV) and Mo(VI) [41]. Our spatially resolved anomalous dispersion analysis (SpReAD) complemented this finding by assigning three of the seven iron ions (Fe1, Fe3, and Fe7) as more reduced than the other four (Fe2, Fe4, Fe5, and Fe6). Assuming a formal oxidation state of Fe(II) for the more reduced sites and Fe(III) for the oxidized positions, this could be integrated with data from 2002 by Noodleman and co-workers, who used broken-symmetry DFT to find that an arrangement of the high-spin metal centers that maximizes antiferromagnetic coupling was the most stable [42]. Their top broken-symmetry solution, BS7, combined with Serena DeBeer’s Mo(III) and our distribution of Fe oxidation states yielded a total spin state of S = 3/2, in line with spectroscopic data (vide infra). I did like this result so much that it almost made me forget that once more, we did not gain much insight into nitrogenase function, staring anxiously at a resting state that still refused to reveal any details of its interaction with N2.
5. Family Business: The Three Nitrogenase Isoforms
Nitrogenase was initially characterized as a molybdenum-containing enzyme [43], but it soon became apparent that it differed from all other known Mo-dependent enzymes in that it did not contain an organic molybdopterin cofactor but had a molybdenum ion as part of an iron–sulfur-based metal cluster (Figure 4a). Only in 1980 did Paul Bishop present evidence for an alternative nitrogen fixation system that was dependent on vanadium [44], and with Bob Eady, he showed that this system had its own structural genes and isolated the enzyme in 1986 [45], as did Hales and co-workers [46]. In 1988, Bishop discovered that a third variant of the nitrogenase system could be isolated from a ΔnifHDK strain, whose activity did not depend on any metal other than iron [47]. As finally confirmed by its genome sequence [48], the model diazotroph A. vinelandii thus contained three different isoforms of nitrogenase that are closely related in structure and function but show variations that allow for important conclusions regarding the overall mechanism. All three nitrogenase isoforms consist of two component proteins. The Fe proteins NifH, VnfH, and AnfH are the dinitrogenase reductases that bind and hydrolyze 2 ATP for each electron transferred via a [4Fe:4S] cluster situated at the interface of the 60 kDa homodimer. The dinitrogenases or MFe proteins (with M = Mo, V, Fe) are built around a heterotetrameric D2K2 core that, in the case of VFe protein and FeFe protein, is extended by two copies of a G subunit. All dinitrogenases contain an electron-transferring [8Fe:7S] P-cluster at the interface of the structurally related D and K subunits, and an active-site cofactor buried deeply within the D subunits (Figure 4). This cofactor is the enigmatic site of N2 reduction, and while the general expectation was that in the isoenzymes, the apical Mo ion of FeMo cofactor was replaced by V or Fe, respectively, any changes in the cofactor environment that might account for the observed differences in substrate specificity and reactivity were unknown. The two alternative nitrogenases are considerably less stable than Mo-nitrogenase and are only produced in the absence of the respective heterometals: Mo repressed V- and Fe-nitrogenase, and V repressed the Fe-dependent enzyme. Our initial attempts at crystallizing a tagged VFe protein from a ΔnifHDK strain of A. vinelandii in collaboration with Markus Ribbe and Yilin Hu were unsuccessful, so we decided to optimize the growth conditions for native A. vinelandii to maximize the production of V-nitrogenase. This process and the subsequent crystallization of the enzyme proved highly challenging [49], and it took more than four years of diligent laboratory work and perseverance by Daniel Sippel to solve the structure of VFe protein and be rewarded with true atomic resolution and unexpected insights into the structural details of the enzyme (Figure 4b). First, we noted that the expected replacement of Mo by V was not the only change at the FeV cofactor. In addition, one of the µ2-bridging sulfide ions at the cluster belt was replaced by a divalent carbonate anion, whose origin and specific role remain unknown [50]. A second finding in VFe protein was far more consequential for understanding nitrogenase catalysis: In addition to the constitutive replacement of one sulfide by carbonate, another µ2-sulfide, termed S2B and bridging Fe2 and Fe6 of the cofactor, was labile and replaced to varying degrees by a single light atom [51]. This binding corresponded to the one observed for the inhibitor CO (vide infra), but the smaller, monoatomic ligand now allowed for the side chain of a conserved glutamine residue near the active site to flip, opening on one side a holding position for the released sulfide S2B and forming a short hydrogen bond to a similarly conserved histidine residue right above the Fe2-Fe6 edge of the cluster. Interestingly, the histidine residue is suggested to serve as a proton source during N2 reduction and is connected to the protein surface via a tight network of hydrogen bonds. For the subsequent integration of a mechanism, the blocking of this proton source when a small ligand binds to the cofactor is essential.
The two key lessons from this structure were that both the active-site cofactor and its surroundings are structurally flexible and that sulfide S2B can be reversibly replaced by a bridging light atom. Following a tradition, we assigned this light atom as a nitrogen species, NH, implying that it might be an intermediate of N2 reduction [51]. Shortly thereafter, this was contested based on a re-analysis of our electron density map [52] and DFT calculations [53] that both favored OH, a bridging hydroxyl. In this case, however, the debate is misleading. The key aspect of replacing sulfide S2B is that it creates a coordination site for substrates and—as we will see—inhibitors that were crucially absent in the resting state of the cofactor. The light atom may be a nitrogen species, it may be oxygen (although this should not originate from O2), or it may even be a carbon species, as it should constitute an intermediate of the respective reduction reaction catalyzed by the enzyme. We do not yet understand why this conformation was seen in V-nitrogenase but so far not in a Mo-nitrogenase, but further support for the key role of this position in catalysis recently came from our structural analysis of the third isoform, Fe-nitrogenase. Extending on our strategy of metal depletion developed to produce V-nitrogenase, we worked with Mo- and V-depleted cultures of A. vinelandii to induce the expression of the anf genes encoding for this enzyme. The three nitrogenases decrease in their activity Mo > V > Fe, and this also correlates to a reduction in both their stability and sensitivity to O2. It was no small feat, and another 4 ½ years of work, that Christian Trncik succeeded in isolating, characterizing, and crystallizing both the Fe protein AnfH [54] and the nitrogenase FeFe protein (Figure 4c) [55]. Fe- and V-nitrogenases of A. vinelandii are more closely related to each other than to the Mo-dependent enzyme, and while the active site held the expected [8Fe:9S:C]:homocitrate FeFe cofactor, it showed a dual conformation of a resting and a turnover state with partial replacement of sulfide S2B that we had also observed in VFe protein [56]. The determination of a three-dimensional structure is always only one aspect of understanding the intricacies of a macromolecular machine, but when solving the structures of the two alternative nitrogenases, I was reminded of the appeal of having this singular moment of discovery when looking at a new electron density map for the first time. All the new information in a structure is revealed at the push of a button, and one can spend time rummaging through its complexity for many hours, discovering fundamental principles and unknown details, always ready to stumble across a surprise around the next turn.
6. Alternative Substrates: Learning from CO about N2
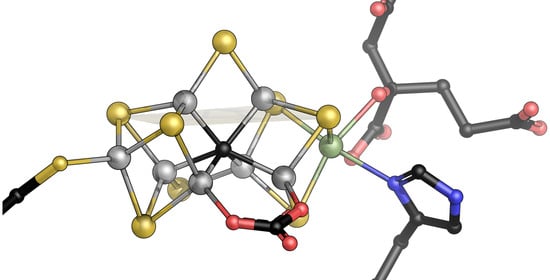
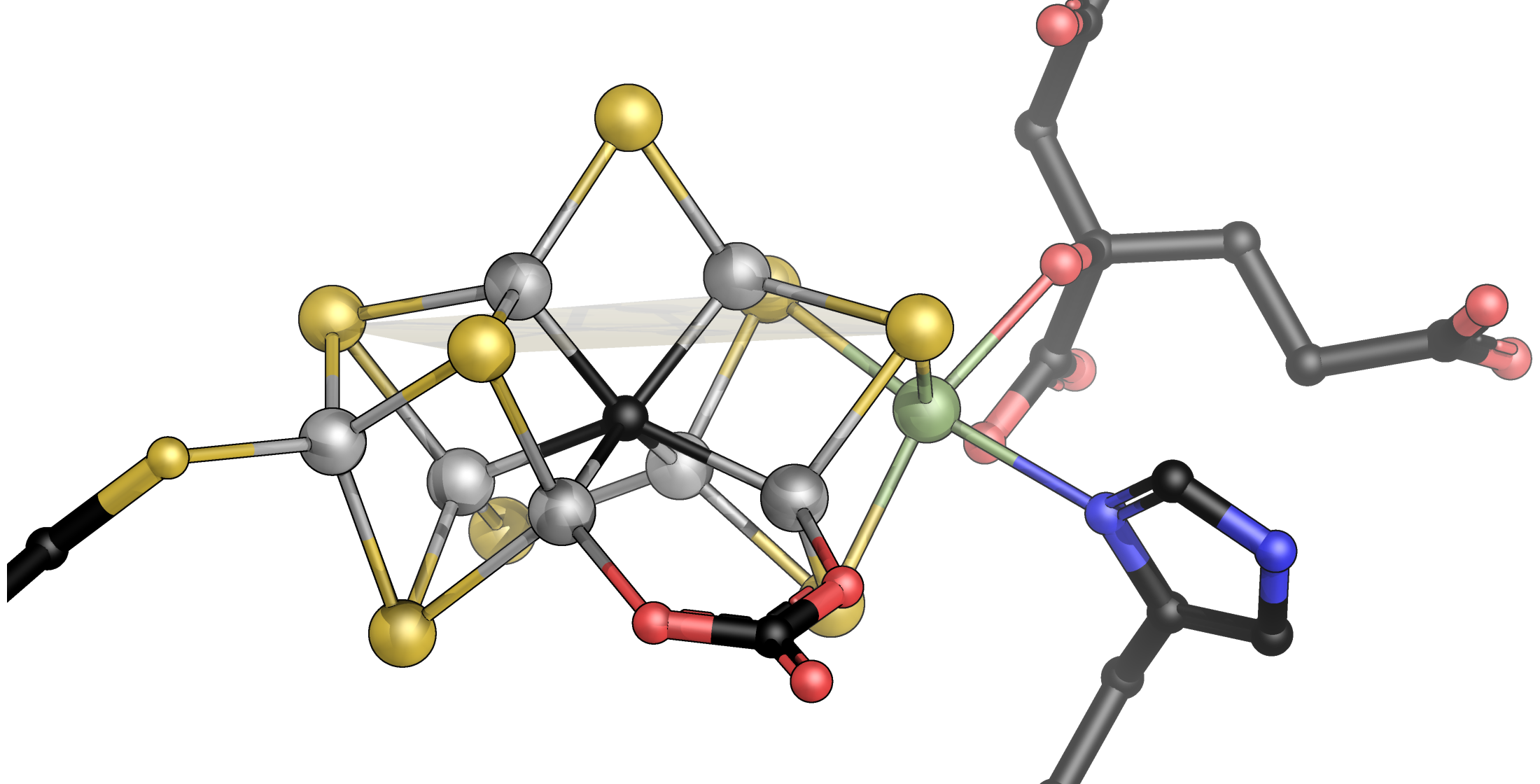
The reduction of N2 is the most challenging and unique catalytic ability of the nitrogenase enzymes. The Lowe–Thorneley scheme dictates that four electrons must be accumulated on the cofactor, and the accumulation of surface hydrides suggested by Seefeldt, Hoffman, and Dean would then allow for the reductive elimination of H2 that leaves the cofactor in a super-reduced state representing the catalytically active species [57,58,59]. The difficulty of even reaching this critical E4 state is highlighted by the high rate of H2 production by nitrogenase—at least in vitro—that represents an abortive side reaction when a surface hydride is accidentally protonated, and two of the accumulated electrons are lost (Figure 3) [60]. All intermediate states on the way from the resting state to E4 are unstable, and the super-reduced E4* state after H2 elimination clearly is highly elusive, although its fingerprints can be seen, for instance, in deuterium exchange experiments [61,62,63]. Turning a kinetic scheme into a mechanism requires adding precise structural information to each intermediate state, and for nitrogenase, this has been a critical point for a long time. States are short-lived, and hydrogen atoms and hydrides cannot easily be depicted by crystallography. The available theoretical models diverge about the electronic properties of the cofactor and, with that, the sites of substrate/intermediate binding. Alternative substrates of the enzyme, however, are less stable than the N2 molecule and may thus be easier to investigate. The most prominent of these alternatives is acetylene, C2H2, a triple-bonded gas isoelectronic to N2 that is reduced to ethylene, C2H4, in a 2-electron reduction reaction that is used as a common assay using a gas chromatograph [64]. Acetylene reacts with nitrogenase already in the E2 state that should be far more accessible than E4, but the properties of this reaction are not ideal: Acetylene is not reduced completely to methane, which would be analogous to N2 reduction, and the carbon atoms are and remain protonated so that the molecule typically interacts with metals side-on via its nucleophilic triple bond [65]. A closer analog to N2 is CO, also with the same number of electrons and high stability, but characterized early on as a non-competitive inhibitor for all known nitrogenase substrates other than protons [66]. This means that its binding diverts all electron flow toward ‘H2 production’, i.e., the protonation of hydrides. ‘Non-competitive’ implies inhibitor binding to the enzyme independent of the substrate. It is mostly found in allosteric mechanisms but also applies if the actual substrate binding site is in a different state for inhibitor binding than it must be to interact with substrates: E2 for CO vs. E4 for N2. As an ideal σ-donor and π-acceptor, the strong-field ligand CO binds strongly to many metal sites so that its inhibitory effect on nitrogenase was far less surprising than the finding by Markus Ribbe and Yilin Hu, who reported in 2010 that the alternative, vanadium-containing nitrogenase system reduces CO [67]. Most interestingly, they found that the dominant (>93%) product of the reaction is not fully reduced methane, CH4, but rather ethylene, as in the case of acetylene reduction [68]. Thus, the enzyme binds CO and is inhibited by it, but it also has a pathway of activating and reducing the molecule, which involves a C-C bond formation and the release of an unsaturated product. Coincidentally, the product range of this reaction is strongly reminiscent of the industrial Fischer–Tropsch process, which, in terms of catalyst and reaction conditions, is analogous to Haber–Bosch nitrogen fixation. In both cases, it is a particular lattice plane of the crystalline iron catalyst that features the exact interatomic spacings to drive the reaction to the observed outcome [69]. In the enzyme, the observation of C-C coupling from two CO molecules implies two distinct binding sites, which was corroborated by decades of spectroscopic studies that showed states designated ‘low-CO’ and ‘high-CO’, with one molecule of CO binding to the enzyme in the former and at least two in the latter [65,70,71,72,73,74,75,76]. In 2014, my former graduate student Thomas Spatzal, now a postdoc with Doug Rees, then succeeded to inhibit Mo-nitrogenase with CO under turnover conditions, isolate MoFe protein from the mixture, grow crystals, and collect diffraction data to 1.5 Å resolution [77]. I vividly remember that when Doug Rees sent me this structure, I was teaching at the 2014 Penn State Workshop on Bioinorganic Chemistry, and I was immediately struck with what I saw because it just looked so … right. This was the very first observation of a ligand bound to a nitrogenase cofactor, and it was nothing like the distorted clusters or surface associations that were predicted, anticipated, or feared. Instead, the CO molecule had ejected the bridging sulfide S2B that later gained prominence in V- and Fe-nitrogenase. CO replaced this atom, forming a bridging carbonyl at Fe2 and Fe6, a classic in metalloorganic chemistry (Figure 5). Michael Rohde and Katharina Parison (née Grunau) in my group did the same for V-nitrogenase and found the exact same binding mode, which was not obvious considering that only V-nitrogenase can reduce this gas [78]. Having a suitable picture of the low-CO state, attempts at describing the high-CO state were made by pressurizing crystals of the CO-complex with more CO, and again, these were successful for MoFe and VFe protein and were reported in parallel by the Rees group and by us [79,80]. In both cases, the binding of a second molecule of CO was observed, this time as a terminal ligand to Fe6, directly adjacent to the first bridging CO molecule of the low-CO state. The two carbon atoms were at a close distance, and the story of C-C bond formation from this geometry literally wrote itself (Figure 5).
It did so, of course, with a few caveats. One was to clarify what states these CO adducts represented. CO does not bind to the resting state of nitrogenase but only requires the enzyme to reach E2 for binding and not E4, as is required for N2 reduction. But what is the actual inhibited state? Discussing this question in our group, many of the countless, isolated data points that were amassed on nitrogenase over decades started to fall into place. This story is and remains a hypothesis but integrates the vast majority of what we know [80]. It goes as follows: The nitrogenase cofactors can only be reduced by a single electron. The second, in E2, already forms the first hydride on the cluster surface. From our structural data, we have proposed that this hydride forms a bridge between Fe2 and Fe6, essentially replacing sulfide S2B (Figure 5) [81]. Theory agrees that S2B is protonated in E1, and one of its bonds to iron is substantially weakened or broken [82], but the calculations so far do not show a complete dissociation of the resulting dangling thiol. It is this hydride-bound E2 state that CO can access, and with the µ-bridging position (that we designate the ‘µ-site’) occupied, the first encounter of CO with the enzyme will be through terminal binding to Fe6 (the ‘t-site’). This complex, then, is where the role of CO is decided. CO can remain bound to the t-site without interacting with the µ-hydride. This will keep the catalytic cycle from progressing forward, and as a result the hydride will eventually be subject to protonation and will be lost as H2. The CO ligand in the t-site then is in the ideal position to migrate to the µ-site and form the bridging carbonyl intermediate seen in the crystal structures [77,78]. Importantly, however, this occurs after release of H2 from the E2 state of the enzyme. The two electrons accumulated to this point are lost, and the enzyme is formally in the resting state E0, but with a bound CO ligand (Figure 5). The low-CO state therefore is an inhibited resting state. It is off-pathway for substrate reduction and requires turnover conditions to return to a catalytically competent state [80,81]. Why then is H2 production not inhibited by CO? We think that along the same lines, the first interaction of any substrate is to form a terminal ligand at Fe6, the t-site. This is also true if the substrate is a proton and the formed intermediate is a terminal hydride [81]. Terminal hydrides are less stable than bridging ones, and this t-hydride would quickly migrate into the µ-site to form the E2 state as described above. In the inhibited state, however, this is prevented by the CO ligand so that the t-hydride is locked in place and will eventually be protonated to form H2. Protons are the only substrate of the enzyme that forms a reactive intermediate at the t-site and can, therefore, still be reduced even if the µ-site is occupied. All other substrates must migrate into the µ-site after initially binding to the t-site for reduction to occur. The µ-site is occupied by a hydride so that the substrate inserts into the bound hydride, resulting in a concerted two-electron reduction. If this substrate is a t-CO, this is what makes the difference between CO inhibition (loss of the hydride as H2) and CO reduction (insertion of CO into the bound µ-hydride, Figure 6). In our proposal, we suggested that from here, two further electron transfers from Fe protein generate another t-hydride at Fe6 that then, in turn, inserts into the bound intermediate so that all reductive steps are two-electron transfers [80]. We cannot exclude that for some or all substrates, it is energetically favorable to directly reduce the bound intermediates rather than form t-hydrides, but this does not affect the essence of the mechanism. The action of nitrogenase thus comes down to a strikingly simple, repetitive two-electron transfer mechanism: A t-hydride is formed and inserted into the µ-site. After four electrons are transferred, the C-O bond is cleaved, water is released, and a methyl group remains bound to the cofactor (Figure 6) [81]. In Fe-nitrogenase, which also reduces CO, this ligand is predominantly protonated and released as methane, while in V-nitrogenase, it stays bound. The reasons for this difference are unclear. In the presence of CO, the VFe protein methyl adduct can then bind another t-CO, which will be inserted into the methyl group, resulting in the enigmatic C-C coupling step. The following two reduction cycles then take this intermediate to bound ethane, which VFe protein again is reluctant to release so that a small amount of longer chain hydrocarbons is formed. For the ethane intermediate, however, the presence of a β-carbon now opens the possibility of a reductive β-hydride elimination to release the product ethylene, leaving a µ-hydride at the cofactor, corresponding to the E2 state [80,81].
The proposed action of the enzyme is straightforward, and a terminal hydride as the active reducing species also has the high reducing power (low ΔE) that makes this enzyme unique. Nevertheless, nitrogenase is the enzyme of biological nitrogen fixation, so how does this apply to N2 reduction that does not require the binding of two molecules of N2 to the enzyme and where there is no N-N bond formation (quite the contrary)? How do these processes differ, and is the discussion above at all relevant? The known part is that the E2 state I described above is not sufficient to activate the inert N2 molecule. The elementary steps of nitrogenase catalysis imply that as the enzyme progresses to E4, a second hydride is formed at the t-site [60]. As this second hydride migrates to the µ-site, it might trigger the elimination of H2 immediately, leaving the cofactor in a two-electron reduced state that now is sufficiently reducing to break the N2 triple bond [51]. However, we disfavor this direct elimination of H2, as the resulting intermediate would be highly reactive and too unstable to persist until substrate N2 diffuses to the cluster and binds. Instead, taking inspiration from model chemistry by Pat Holland [83], Jonas Peters [84], and others, we suggest that the second hydride forms a bis-µ2-hydride diamond core with Fe2 and Fe6, possibly triggered by the binding of a N2 molecule to the t-site. The two adjacent hydrides then eliminate H2, with N2 in a perfect position for direct reduction as it migrates into the µ-site [55,60]. This one point where the reductive elimination of H2 is used as a catalytic trick to generate a 2-electron-reduced cofactor is the only part of the reaction that is unique to N2 fixation. With the triple bond broken, the resulting intermediates can be reduced in full analogy to the steps for CO reduction outlined above, and product NH4+ is eventually released.
These steps also suggest structures of intermediates, some of which have already been defined by spectroscopy [82,85,86]. It will be very challenging to visualize these intermediates even at the high resolution of current crystal structures, but I emphasize that this mechanism for nitrogenase is not merely the fever dream of a desperate structural biologist. It gains its value from integrating very well with almost all existing data and rationalizes mechanistic peculiarities that have, in some cases, been known for decades. Of course, it also leaves us with open questions and thus highlights the specific points where further study is needed.
7. So What about Molybdenum?
Ralf Mendel deserves high praise for putting together this series of essays centered on molybdenum in the living world, and I realize that while the Mo ion in nitrogenase is unique in biology [87], little has been said about its value in the enzyme, other than that some things actually work better without it. Nevertheless, although its biogenesis is the most intricate of the three isoenzymes, Mo-nitrogenase presumably is the evolutionarily oldest one, and it also is the one that all diazotrophs possess. Biological nitrogen fixation dates back far in evolution [88,89] and is found in diazotrophs of the kingdoms of bacteria and archaea but not in eukaryotes. The switch to a V-dependent enzyme may have been triggered by the transition of organisms from marine habitats, where molybdate is quite abundant, to terrestrial ones, where Mo availability is lower, while vanadate presents an alternative. Another hypothesis is that the differences in catalytic abilities, and foremost the reactivity toward CO, may be of physiological relevance so that the alternative enzymes are not (or not only) intended for nitrogen fixation [90]. For a long time, and once in a while still today, the Mo ion of FeMo cofactor was also suspected to be the actual binding site for substrates [91,92], and the possibly labile, organic homocitrate ligand at this site could dissociate to open up a binding site. Current data favor iron, as discussed above, but with nitrogenase, one should always be ready to be surprised.
The role of molybdenum thus remains enigmatic, but a closer look at the current model for the electronic structure of the cofactors may provide some hints (Figure 7). The coupling of the high-spin metal centers in the FeMo cofactor is best described by the BS7 coupling scheme [93]. For Fe2 and Fe6, the iron ions involved in substrate binding, this scheme likely establishes antiferromagnetic coupling to all surrounding metals—with one exception. Fe2 couples ferromagnetically with the apical Fe1, and Fe6 does so with the other apex of the cofactor that in Mo- and V-nitrogenase is occupied by the heterometal. In the preceding discussion, I have presented Fe6 as the key player for substrate binding to nitrogenase—a postulate that Dos Santos and colleagues first made based on EPR data from a variant MoFe protein—and this is the exact site that couples most strongly to the heterometal [94]. The electronic structures at this point are more complicated, but even if Mo (or V) are not the metals that bind dinitrogen, their influence on the reactivity of the overall process is immediate and important. The surrounding of the cofactors is strikingly similar in all three isoforms, and the residues in the cluster cavity are fully conserved. It is reasonable and more than likely that many of the observed differences are directly due to the influence of the heterometal. Another interesting implication of this model is that the large multi-metal cofactors only use two iron ions for substrate and hydride formation. What then explains the size of the cofactor and the investment into its biogenesis? Why are there no simpler dinuclear metal sites able to catalyze N2 fixation? Looking once more at model complexes, we can hypothesize that an E4 state with two bridging hydrides as in the Holland and Peters models [83,84] may even be too stable to efficiently eliminate E2 if allowed to relax its metal–metal distance. In the nitrogenase cofactors, the unique Fe6:µ6-C core with the other µ2 bridging ligands in place will restrain the Fe2–Fe6 distance, destabilizing the E4 state to favor efficient H2 elimination. I have described this carbon-doted iron core of the cofactor as its ‘heart of steel’ to emphasize the relevance of its rigidity, which has been favorably taken up by the community [95].
8. What Is Next in Nitrogenase Research?
Necessarily, this review provided a limited perspective on a large, highly complex, and long-worked field of research. I have focused on the level of understanding that we have gained by studying the structures, properties, and interactions of the three known isoforms of nitrogenase, Mo-, V-, and Fe-dependent enzyme systems, seen through the focus of the contributions of my team. The mechanistic hypotheses outlined here are an attempt at integrating as much of the available data as possible, but they do leave some open questions of their own. We do not yet have a suitable understanding of the differences in the reactivity of the isoenzymes. Many of the postulated intermediates could be drawn terminal to Fe2 (as we did) or bridging both irons and the older hypothesis that the first surface hydride forms in the E2 state was recently challenged for FeFe protein, where a hydride was detected in E1, implying an oxidation of the cofactor [96]. All these points will be addressed, and the field is eagerly awaiting the theory to mature into a reliable arbiter with predictive power for complex metal clusters. An enzyme such as nitrogenase, of course, deserves attention along many other lines of research: The evolutionary history of nitrogen fixation is enigmatic and fascinating, with its origin tracing back to—of all things—enzymes from ancient tetrapyrrole biogenesis pathway, including those for bacteriochlorophyll [97] and for coenzyme F430, the unique nickel porphyrin cofactor of methanogenic archaea [98,99]. The relationship and evolution of the extant nitrogenases have inspired attempts at reconstructing ancient nitrogenases through reverse phylogenetic engineering [100], and the Fe proteins as ATP-driven low-potential reductases have been found in a variety of other contexts, including challenging radical reactions [101]. The interplay of Fe proteins and dinitrogenases also goes far beyond a simple electron transfer reaction, and the mechanisms that convert the chemical energy of ATP phosphodiester bond hydrolysis into a lowered midpoint potential of the electron that reaches the active-site cofactor are a busy field of study [102,103]. Another vast area of nitrogenase research is the biogenesis of the enzymes and their metal clusters. The assembly of MoFe protein requires approximately 20 gene products [104], although production of the enzyme from a minimal gene cluster has been reported [105], and even the less intricate Fe-nitrogenase is dependent on a minimal set of nine genes when choosing a suitable expression host [106]. For the dinitrogenase, the P-clusters are inserted into the apoprotein as a pair of [4Fe:4S] clusters and then fused through the action of the Fe protein [25], while the cofactor is assembled ex situ through several states and only then inserted into the enzyme [107]. It is due to this complexity that, to this day, there is no convenient heterologous production system for nitrogenase, although an excellent genetic system for homologous protein engineering in A. vinelandii has been established by Dennis Dean and co-workers and is widely in use today [108,109].
Many questions thus remain to be answered, but although this challenge has been compared to the first ascent to a mountain summit [110], the motivation here is not just that ‘it’s there’. Fixing atmospheric nitrogen is a metabolic ability that emerged early in evolution but has never made it into the eukaryotic world. Nitrogen availability quickly becomes a growth-limiting factor if biomass is removed from an environment, i.e., in any modern agricultural setting. This was where the Haber–Bosch process was a game-changer [111] that led to a present where half of the human population can only be sustained through the use of nitrogen fertilizers [112]. Fertilizer use leads to nitrogen pollution, whose mitigation poses severe challenges [113], and an obvious solution is to transfer the ability to use atmospheric N2 as a nitrogen source for growth into staple crops. Major efforts are spent to produce active nitrogenase in plant mitochondria [114,115,116], and there is no doubt that in order to succeed, we need a well-founded and detailed understanding of the assembly, action, and regulation of this intriguing enzyme system. The story of nitrogenase is far from told.
Funding
This research was funded by the European Research Council (grant no. 310656) and Deutsche Forschungsgemeinschaft (CRC 1381, project ID 403222702; PP 1927, project ID 311061829; and RTG 1976, project ID 235777276).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
As a group leader, I am lucky and privileged to have worked and work today with a series of outstanding young scientists as graduate students and postdocs on all our proteins of interest. While I have mentioned some key actors in the text, my nitrogenase team has benefited from the work of many more, and I express my thanks to Haitham Saad Eddin, Thomas Spatzal, Eva-Maria Burger, Ivana Djurdjevic, Laure Decamps, Julia Netzer, Daniel Sippel, Michael Rohde, Christian Trncik, Jakob Gies-Elterlein, Florian Schneider, Lin Zhang, Philipp Franke, Katharina Parison, and Franka Detemple, as well as to the many B.Sc. and M.Sc. students that worked with them. I also acknowledge the work of Stefan Gerhardt and Lorenz Heidinger and the technical assistance of Toni Espin and Sandra Würstlin. The nitrogenase field is as diverse as bioinorganic chemistry itself, reaching into many disciplines of natural sciences. Success in this field can only come from contributing to a network of collaborations with excellent colleagues around the globe. I am deeply grateful for the mentorship and friendship of Frank Neese, Peter Kroneck, Albrecht Messerschmidt, Robert Huber, Doug Rees, Jim Howard, and Ralf Ficner. Over the years, we have profited from collaborations and discussions with Susana Andrade, Holger Dobbek, Akif Tezcan, Markus Ribbe, Yilin Hu, Patricia dos Santos, Dennis Dean, Brian Hoffman, Lance Seefeldt, Betül Kaçar, Pat Holland, Franc Meyer, Ragnar Björnsson, Shelley Minteer, Anna Fischer, Ingo Zebger, Sven Stripp, and Volker Schünemann.
Conflicts of Interest
The author declares no conflict of interest.
References
- Hellriegel, H.; Willfarth, H. Untersuchungen über die Stickstoffnährung der Gramineen und Leguminosen. Beilageheft Z. Ver. Rübenzucker-Ind. Dtsch. Reichs 1888, 38, 1–234. [Google Scholar]
- Lipman, J.G. Experiments on the transformation and fixation of nitrogen by bacteria. Rep. N. J. Agric. Exper. Stat. 1903, 24, 217–285. [Google Scholar]
- Canfield, D.E.; Glazer, A.N.; Falkowski, P.G. The Evolution and Future of Earth’s Nitrogen Cycle. Science 2010, 330, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Smil, V. Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production; MIT Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Bagchi, S.N.; Ernst, A.; Böger, P. The Effect of Activated Oxygen Species on Nitrogenase of Anabaena variabilis. Z. Naturforsch. C 1991, 46, 407–415. [Google Scholar] [CrossRef]
- Kim, J.S.; Rees, D.C. Structural Models for the Metal Centers in the Nitrogenase Molybdenum-Iron Protein. Science 1992, 257, 1677–1682. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Rees, D.C. Crystallographic Structure and Functional Implications of the Nitrogenase Molybdenum Iron Protein from Azotobacter vinelandii. Nature 1992, 360, 553–560. [Google Scholar]
- Georgiadis, M.M.; Komiya, H.; Chakrabarti, P.; Woo, D.; Kornuc, J.J.; Rees, D.C. Crystallographic Structure of the Nitrogenase Iron Protein from Azotobacter vinelandii. Science 1992, 257, 1653–1659. [Google Scholar] [CrossRef]
- Bulen, W.A.; LeComte, J.R. Nitrogenase System from Azotobacter: 2-Enzyme Requirement for N2 Reduction, ATP-Dependent H2 Evolution and ATP Hydrolysis. Proc. Natl. Acad. Sci. USA 1966, 56, 979–986. [Google Scholar] [CrossRef]
- Deisenhofer, J.; Epp, O.; Miki, K.; Huber, R.; Michel, H. Structure of the Protein Subunits in the Photosynthetic Reaction Center of Rhodopseudomonas viridis at 3 Å Resolution. Nature 1985, 318, 618–624. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 22. [Google Scholar] [CrossRef]
- Messerschmidt, A.; Rossi, A.; Ladenstein, R.; Huber, R.; Bolognesi, M.; Gatti, G.; Marchesini, A.; Petruzzelli, R.; Finazziagro, A. X-Ray Crystal-Structure of the Blue Oxidase Ascorbate Oxidase from Zucchini—Analysis of the Polypeptide Fold and a Model of the Copper Sites and Ligands. J. Mol. Biol. 1989, 206, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, A.; Steigemann, W.; Huber, R.; Lang, G.; Kroneck, P.M.H. X-Ray Crystallographic Characterization of Type-2-Depleted Ascorbate Oxidase from Zucchini. Eur. J. Biochem. 1992, 209, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.K.; Mukund, S.; Kletzin, A.; Adams, M.W.W.; Rees, D.C. Structure of a Hyperthermophilic Tungstopterin Enzyme, Aldehyde Ferredoxin Oxidoreductase. Science 1995, 267, 1463–1469. [Google Scholar] [CrossRef]
- Romao, M.J.; Archer, M.; Moura, I.; Moura, J.J.G.; LeGall, J.; Engh, R.; Schneider, M.; Hof, P.; Huber, R. Crystal-Structure of the Xanthine Oxidase-Related Aldehyde Oxidoreductase from D. gigas. Science 1995, 270, 1170–1176. [Google Scholar] [CrossRef]
- Einsle, O.; Messerschmidt, A.; Stach, P.; Bourenkov, G.P.; Bartunik, H.D.; Huber, R.; Kroneck, P.M.H. Structure of cytochrome c nitrite reductase. Nature 1999, 400, 476–480. [Google Scholar] [CrossRef]
- Peters, J.W.; Stowell, M.H.B.; Soltis, S.M.; Finnegan, M.G.; Johnson, M.K.; Rees, D.C. Redox-dependent structural changes in the nitrogenase P-cluster. Biochemistry 1997, 36, 1181–1187. [Google Scholar] [CrossRef]
- Einsle, O.; Tezcan, F.A.; Andrade, S.L.A.; Schmid, B.; Yoshida, M.; Howard, J.B.; Rees, D.C. Nitrogenase MoFe-protein at 1.16 Å resolution: A central ligand in the FeMo-cofactor. Science 2002, 297, 1696–1700. [Google Scholar] [CrossRef]
- Smith, B.E. Nitrogenase reveals its inner secrets. Science 2002, 297, 1654–1655. [Google Scholar] [CrossRef]
- Lancaster, K.M.; Roemelt, M.; Ettenhuber, P.; Hu, Y.L.; Ribbe, M.W.; Neese, F.; Bergmann, U.; DeBeer, S. X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [Google Scholar] [CrossRef]
- Spatzal, T.; Aksoyoğlu, M.; Zhang, L.M.; Andrade, S.L.A.; Schleicher, E.; Weber, S.; Rees, D.C.; Einsle, O. Evidence for Interstitial Carbon in Nitrogenase FeMo Cofactor. Science 2011, 334, 940. [Google Scholar] [CrossRef] [PubMed]
- Tezcan, F.A.; Kaiser, J.T.; Mustafi, D.; Walton, M.Y.; Howard, J.B.; Rees, D.C. Nitrogenase complexes: Multiple docking sites for a nucleotide switch protein. Science 2005, 309, 1377–1380. [Google Scholar] [CrossRef]
- Hu, Y.L.; Corbett, M.C.; Fay, A.W.; Webber, J.A.; Hodgson, K.O.; Hedman, B.; Ribbe, M.W. FeMo cofactor maturation on NifEN. Proc. Natl. Acad. Sci. USA 2006, 103, 17119–17124. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Fay, A.W.; Lee, C.C.; Ribbe, M.W. P-cluster maturation on nitrogenase MoFe protein. Proc. Natl. Acad. Sci. USA 2007, 104, 10424–10429. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Fay, A.W.; Lee, C.C.; Yoshizawa, J.; Ribbe, M.W. Assembly of nitrogenase MoFe protein. Biochemistry 2008, 47, 3973–3981. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.M.; Ludden, P.W. Maturation of nitrogenase: A biochemical puzzle. J. Bacteriol. 2005, 187, 405–414. [Google Scholar] [CrossRef]
- Rubio, L.M.; Hernandez, J.A.; Soboh, B.; Zhao, D.; Igarashi, R.Y.; Curatti, L.; Ludden, P.W. The Role of Nif Proteins in Nitrogenase Maturation. Curr. Plant Sci. Biot. 2008, 42, 325–328. [Google Scholar]
- Lukoyanov, D.; Barney, B.M.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. Connecting nitrogenase intermediates with the kinetic scheme for N2 reduction by a relaxation protocol and identification of the N2 binding state. Proc. Natl. Acad. Sci. USA 2007, 104, 1451–1455. [Google Scholar] [CrossRef]
- Yang, T.C.; Maeser, N.K.; Laryukhin, M.; Lee, H.I.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. The interstitial atom of the nitrogenase FeMo-Cofactor: ENDOR and ESEEM evidence that it is not a nitrogen. J. Am. Chem. Soc. 2005, 127, 12804–12805. [Google Scholar] [CrossRef]
- Lukoyanov, D.; Pelmenschikov, V.; Maeser, N.; Laryukhin, M.; Yang, T.C.; Noodleman, L.; Dean, D.R.; Case, D.A.; Seefeldt, L.C.; Hoffman, B.M. Testing if the interstitial atom, X, of the nitrogenase molybdenum-iron cofactor is N or C: ENDOR, ESEEM, and DFT studies of the S=3/2 resting state in multiple environments. Inorg. Chem. 2007, 46, 11437–11449. [Google Scholar] [CrossRef]
- Wiig, J.A.; Hu, Y.L.; Lee, C.C.; Ribbe, M.W. Radical SAM-Dependent Carbon Insertion into the Nitrogenase M-Cluster. Science 2012, 337, 1672–1675. [Google Scholar] [CrossRef]
- Lowe, D.J.; Thorneley, R.N.F. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—Pre-Steady-State Kinetics of H2 Formation. Biochem. J. 1984, 224, 877–886. [Google Scholar] [CrossRef]
- Lowe, D.J.; Thorneley, R.N.F. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—The Determination of Rate Constants Required for the Simulation of the Kinetics of N2 Reduction and H2 Evolution. Biochem. J. 1984, 224, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Thorneley, R.N.F.; Lowe, D.J. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—Pre-Steady-State Kinetics of an Enzyme-Bound Intermediate in N2 Reduction and of NH3 Formation. Biochem. J. 1984, 224, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Thorneley, R.N.F.; Lowe, D.J. The Mechanism of Klebsiella pneumoniae Nitrogenase Action—Simulation of the Dependences of H2 Evolution Rate on Component-Protein Concentration and Ratio and Sodium Dithionite Concentration. Biochem. J. 1984, 224, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Thorneley, R.N.F.; Lowe, D.J. Kinetics and mechanism of the nitrogenase enzyme system. In Molybdenum Enzymes; Spiro, T.G., Ed.; Wiley-Interscience: New York, NY, USA, 1985; Volume 1, pp. 221–284. [Google Scholar]
- Spatzal, T.; Einsle, O.; Andrade, S.L. Analysis of the Magnetic Properties of Nitrogenase FeMo Cofactor by Single-Crystal EPR Spectroscopy. Angew. Chem. 2013, 52, 10116–10119. [Google Scholar] [CrossRef] [PubMed]
- Einsle, O.; Andrade, S.L.; Dobbek, H.; Meyer, J.; Rees, D.C. Assignment of individual metal redox states in a metalloprotein by crystallographic refinement at multiple X-ray wavelengths. J. Am. Chem. Soc. 2007, 129, 2210–2211. [Google Scholar] [CrossRef]
- Spatzal, T.; Schlesier, J.; Burger, E.M.; Sippel, D.; Zhang, L.M.; Andrade, S.L.A.; Rees, D.C.; Einsle, O. Nitrogenase FeMoco investigated by spatially resolved anomalous dispersion refinement. Nat. Commun. 2016, 7, 10902. [Google Scholar] [CrossRef]
- Björnsson, R.; Lima, F.A.; Spatzal, T.; Weyhermüller, T.; Glatzel, P.; Bill, E.; Einsle, O.; Neese, F.; DeBeer, S. Identification of a spin-coupled Mo(III) in the nitrogenase iron-molybdenum cofactor. Chem. Sci. 2014, 5, 3096–3103. [Google Scholar] [CrossRef]
- Lovell, T.; Li, J.; Liu, T.Q.; Case, D.A.; Noodleman, L. FeMo cofactor of nitrogenase: A density functional study of states MN, MOX, MR, and MI. J. Am. Chem. Soc. 2001, 123, 12392–12410. [Google Scholar] [CrossRef]
- Chatt, J.; Dilworth, J.R.; Richards, R.L.; Sanders, J.R. Chemical Evidence Concerning Function of Molybdenum in Nitrogenase. Nature 1969, 224, 1201. [Google Scholar] [CrossRef] [PubMed]
- Bishop, P.E.; Jarlenski, D.M.L.; Hetherington, D.R. Evidence for an Alternative Nitrogen Fixation System in Azotobacter vinelandii. Proc. Natl. Acad. Sci. USA 1980, 77, 7342–7346. [Google Scholar] [CrossRef]
- Bishop, P.E.; Hawkins, M.E.; Eady, R.R. Nitrogen fixation in molybdenum-deficient continuous culture by a strain of Azotobacter vinelandii carrying a deletion of the structural genes for nitrogenase (nifHDK). Biochem. J. 1986, 238, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Hales, B.J.; Case, E.E.; Morningstar, J.E.; Dzeda, M.F.; Mauterer, L.A. Isolation of a New Vanadium-Containing Nitrogenase from Azotobacter vinelandii. Biochemistry 1986, 25, 7253–7255. [Google Scholar] [CrossRef] [PubMed]
- Chisnell, J.R.; Premakumar, R.; Bishop, P.E. Purification of a 2nd Alternative Nitrogenase from a nifHDK Deletion Strain of Azotobacter vinelandii. J. Bacteriol. 1988, 170, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Setubal, J.C.; dos Santos, P.; Goldman, B.S.; Ertesvag, H.; Espin, G.; Rubio, L.M.; Valla, S.; Almeida, N.F.; Balasubramanian, D.; Cromes, L.; et al. Genome Sequence of Azotobacter vinelandii, an Obligate Aerobe Specialized To Support Diverse Anaerobic Metabolic Processes. J. Bacteriol. 2009, 191, 4534–4545. [Google Scholar] [CrossRef]
- Sippel, D.; Schlesier, J.; Rohde, M.; Trncik, C.; Decamps, L.; Djurdjevic, I.; Spatzal, T.; Andrade, S.L.A.; Einsle, O. Production and isolation of vanadium nitrogenase from Azotobacter vinelandii by molybdenum depletion. J. Biol. Inorg. Chem. 2017, 22, 161–168. [Google Scholar] [CrossRef]
- Sippel, D.; Einsle, O. The structure of vanadium nitrogenase reveals an unusual bridging ligand. Nat. Chem. Biol. 2017, 13, 956–960. [Google Scholar] [CrossRef]
- Sippel, D.; Rohde, M.; Netzer, J.; Trncik, C.; Gies, J.; Grunau, K.; Djurdjevic, I.; Decamps, L.; Andrade, S.L.A.; Einsle, O. A bound reaction intermediate sheds light on the mechanism of nitrogenase. Science 2018, 359, 1484–1489. [Google Scholar] [CrossRef]
- Cao, L.L.; Caldararu, O.; Ryde, U. Does the crystal structure of vanadium nitrogenase contain a reaction intermediate? Evidence from quantum refinement. J. Biol. Inorg. Chem. 2020, 25, 847–861. [Google Scholar] [CrossRef]
- Benediktsson, B.; Björnsson, R. Quantum Mechanics/Molecular Mechanics Study of Resting-State Vanadium Nitrogenase: Molecular and Electronic Structure of the Iron-Vanadium Cofactor. Inorg. Chem. 2020, 59, 11514–11527. [Google Scholar] [CrossRef] [PubMed]
- Trncik, C.; Müller, T.; Franke, P.; Einsle, O. Structural analysis of the reductase component AnfH of iron-only nitrogenase from Azotobacter vinelandii. J. Inorg. Biochem. 2022, 227, 111690. [Google Scholar] [CrossRef]
- Trncik, C.; Detemple, F.; Einsle, O. Iron-only Fe-Nitrogenase underscores common catalytic principles in Biological Nitrogen Fixation. Nat. Catal. 2023, 6, 415–424. [Google Scholar] [CrossRef]
- Einsle, O.; Rees, D.C. Structural Enzymology of Nitrogenase Enzymes. Chem. Rev. 2020, 120, 4969–5004. [Google Scholar] [CrossRef] [PubMed]
- Lukoyanov, D.; Khadka, N.; Yang, Z.Y.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. Reductive Elimination of H2 Activates Nitrogenase to Reduce the N-N Triple Bond: Characterization of the E4(4H) Janus Intermediate in Wild-Type Enzyme. J. Am. Chem. Soc. 2016, 138, 10674–10683. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.F.; Lukoyanov, D.A.; Shaw, S.; Compton, P.; Tokmina-Lukaszewska, M.; Bothner, B.; Kelleher, N.; Dean, D.R.; Hoffman, B.M.; Seefeldt, L.C. Mechanism of N2 Reduction Catalyzed by Fe-Nitrogenase Involves Reductive Elimination of H2. Biochemistry 2018, 57, 701–710. [Google Scholar] [CrossRef]
- Harris, D.F.; Lukoyanov, D.A.; Kallas, H.; Trncik, C.; Yang, Z.Y.; Compton, P.; Kelleher, N.; Einsle, O.; Dean, D.R.; Hoffman, B.M.; et al. Mo-, V-, and Fe-Nitrogenases Use a Universal Eight-Electron Reductive-Elimination Mechanism To Achieve N2 Reduction. Biochemistry 2019, 58, 3293–3301. [Google Scholar] [CrossRef]
- Rohde, M.; Sippel, D.; Trncik, C.; Andrade, S.L.A.; Einsle, O. The Critical E4 State of Nitrogenase Catalysis. Biochemistry 2018, 57, 5497–5504. [Google Scholar] [CrossRef]
- Burgess, B.K.; Wherland, S.; Newton, W.E.; Stiefel, E.I. Nitrogenase Reactivity—Insight into the Nitrogen-Fixing Process through Hydrogen-Inhibition and HD-Forming Reactions. Biochemistry 1981, 20, 5140–5146. [Google Scholar] [CrossRef]
- Fisher, K.; Dilworth, M.J.; Newton, W.E. Differential effects on N2 binding and reduction, HD formation, and azide reduction with alpha-195(His)- and alpha-191(Gln)-substituted MoFe proteins of Azotobacter vinelandii nitrogenase. Biochemistry 2000, 39, 15570–15577. [Google Scholar] [CrossRef]
- Harris, D.F.; Yang, Z.Y.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. Kinetic Understanding of N2 Reduction versus H2 Evolution at the E4 (4H) Janus State in the Three Nitrogenases. Biochemistry 2018, 57, 5706–5714. [Google Scholar] [CrossRef] [PubMed]
- Dilworth, M.J. Acetylene Reduction by Nitrogen-Fixing Preparations from Clostridium Pasteurianum. Biochim. Biophys. Acta 1966, 127, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.J.; Eady, R.R.; Thorneley, R.N.F. Electron-Paramagnetic-Resonance Studies on Nitrogenase of Klebsiella pneumoniae—Evidence for Acetylene-Nitrogenase and Ethylene-Nitrogenase Transient Complexes. Biochem. J. 1978, 173, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.C.; Chen, C.H.; Burris, R.H. Inhibition of Nitrogenase-Catalyzed Reductions. Biochim. Biophys. Acta 1973, 292, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Hu, Y.L.; Ribbe, M.W. Vanadium Nitrogenase Reduces CO. Science 2010, 329, 642. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.C.; Hu, Y.L.; Ribbe, M.W. Insights into Hydrocarbon Formation by Nitrogenase Cofactor Homologs. Mbio 2015, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Ertl, G.; Huber, M.; Lee, S.B.; Paal, Z.; Weiss, M. Interactions of Nitrogen and Hydrogen on Iron Surfaces. Appl. Surf. Sci. 1981, 8, 373–386. [Google Scholar] [CrossRef]
- Davis, L.C.; Henzl, M.T.; Burris, R.H.; Orme-Johnson, W.H. Iron-sulfur clusters in the molybdenum-iron protein component of nitrogenase. Electron paramagnetic resonance of the carbon monoxide inhibited state. Biochemistry 1979, 18, 4860–4869. [Google Scholar] [CrossRef]
- Lee, H.I.; Cameron, L.M.; Hales, B.J.; Hoffman, B.M. CO binding to the FeMo cofactor of CO-inhibited nitrogenase: (CO)-13C and 1H Q-band ENDOR investigation. J. Am. Chem. Soc. 1997, 119, 10121–10126. [Google Scholar] [CrossRef]
- Pollock, R.C.; Lee, H.I.; Cameron, L.M.; Derose, V.J.; Hales, B.J.; Orme-Johnson, W.H.; Hoffman, B.M. Investigation of CO Bound to Inhibited Forms of Nitrogenase MoFe Protein by 13C ENDOR. J. Am. Chem. Soc. 1995, 117, 8686–8687. [Google Scholar] [CrossRef]
- George, S.J.; Ashby, G.A.; Wharton, C.W.; Thorneley, R.N.F. Time-resolved binding of carbon monoxide to nitrogenase monitored by stopped-flow infrared spectroscopy. J. Am. Chem. Soc. 1997, 119, 6450–6451. [Google Scholar] [CrossRef]
- Cameron, L.M.; Hales, B.J. Investigation of CO binding and release from Mo-nitrogenase during catalytic turnover. Biochemistry 1998, 37, 9449–9456. [Google Scholar] [CrossRef] [PubMed]
- Maskos, Z.; Hales, B.J. Photo-lability of CO bound to mo-nitrogenase from Azotobacter vinelandii. J. Inorg. Biochem. 2003, 93, 11–17. [Google Scholar] [CrossRef]
- Yan, L.F.; Pelmenschikov, V.; Dapper, C.H.; Scott, A.D.; Newton, W.E.; Cramer, S.P. IR-Monitored Photolysis of CO-Inhibited Nitrogenase: A Major EPR-Silent Species with Coupled Terminal CO Ligands. Chem.-Eur. J. 2012, 18, 16349–16357. [Google Scholar] [CrossRef]
- Spatzal, T.; Perez, K.A.; Einsle, O.; Howard, J.B.; Rees, D.C. Ligand binding to the FeMo-cofactor: Structures of CO-bound and reactivated nitrogenase. Science 2014, 345, 1620–1623. [Google Scholar] [CrossRef] [PubMed]
- Rohde, M.; Grunau, K.; Einsle, O. CO Binding to the FeV Cofactor of CO-Reducing Vanadium Nitrogenase at Atomic Resolution. Angew. Chem. Int. Edit. 2020, 59, 23626–23630. [Google Scholar] [CrossRef]
- Buscagan, T.M.; Perez, K.A.; Maggiolo, A.O.; Rees, D.C.; Spatzal, T. Structural Characterization of Two CO Molecules Bound to the Nitrogenase Active Site. Angew. Chem. Int. Edit. 2021, 60, 5704–5707. [Google Scholar] [CrossRef]
- Rohde, M.; Laun, K.; Zebger, I.; Stripp, S.T.; Einsle, O. Two ligand-binding sites in CO-reducing V nitrogenase reveal a general mechanistic principle. Sci. Adv. 2021, 7, eabg4474. [Google Scholar] [CrossRef]
- Einsle, O. Catalysis and structure of nitrogenase. Curr. Opin. Struct. Biol. 2023, 83, 102719. [Google Scholar] [CrossRef]
- Van Stappen, C.; Thorhallsson, A.T.; Decamps, L.; Björnsson, R.; DeBeer, S. Resolving the structure of the E1 state of Mo nitrogenase through Mo and Fe K-edge EXAFS and QM/MM calculations. Chem. Sci. 2019, 10, 9807–9821. [Google Scholar] [CrossRef]
- Smith, J.M.; Lachicotte, R.J.; Holland, P.L. N=N bond cleavage by a low-coordinate iron(II) hydride complex. J. Am. Chem. Soc. 2003, 125, 15752–15753. [Google Scholar] [CrossRef] [PubMed]
- Rittle, J.; McCrory, C.C.L.; Peters, J.C. A 106-Fold Enhancement in N2-Binding Affinity of an Fe2(µ-H)2 Core upon Reduction to a Mixed-Valence FeIIFeI State. J. Am. Chem. Soc. 2014, 136, 13853–13862. [Google Scholar] [CrossRef] [PubMed]
- Van Stappen, C.; Davydov, R.; Yang, Z.Y.; Fan, R.; Guo, Y.; Bill, E.; Seefeldt, L.C.; Hoffman, B.M.; DeBeer, S. Spectroscopic Description of the E1 State of Mo Nitrogenase Based on Mo and Fe X-ray Absorption and Mössbauer Studies. Inorg. Chem. 2019, 58, 12365–12376. [Google Scholar] [CrossRef]
- Van Stappen, C.; Decamps, L.; Cutsail, G.E.; Bjornsson, R.; Henthorn, J.T.; Birrell, J.A.; DeBeer, S. The Spectroscopy of Nitrogenases. Chem. Rev. 2020, 120, 5005–5081. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.K.; Ugalde, R.A.; Imperial, J.; Brill, W.J. Molybdenum in Nitrogenase. Annu. Rev. Biochem. 1984, 53, 231–257. [Google Scholar] [CrossRef] [PubMed]
- Boyd, E.S.; Costas, A.M.G.; Hamilton, T.L.; Mus, F.; Peters, J.W. Evolution of Molybdenum Nitrogenase during the Transition from Anaerobic to Aerobic Metabolism. J. Bacteriol. 2015, 197, 1690–1699. [Google Scholar] [CrossRef]
- Pi, H.W.; Lin, J.J.; Chen, C.A.; Wang, P.H.; Chiang, Y.R.; Huang, C.C.; Young, C.C.; Li, W.H. Origin and Evolution of Nitrogen Fixation in Prokaryotes. Mol. Biol. Evol. 2022, 39, 9. [Google Scholar] [CrossRef]
- Rebelein, J.G.; Lee, C.C.; Hu, Y.L.; Ribbe, M.W. The in vivo hydrocarbon formation by vanadium nitrogenase follows a secondary metabolic pathway. Nat. Commun. 2016, 7, 13641. [Google Scholar] [CrossRef]
- Durrant, M.C. A molybdenum-centred model for nitrogenase catalysis. Inorg. Chem. Commun. 2001, 4, 60–62. [Google Scholar] [CrossRef]
- Coucouvanis, D.; Mosier, P.E.; Demadis, K.D.; Patton, S.; Malinak, S.M.; Kim, C.G.; Tyson, M.A. The Catalytic Reduction of Hydrazine to Ammonia by the MoFe3S4 Cubanes and Implications Regarding the Function of Nitrogenase—Evidence for Direct Involvement of the Molybdenum Atom in Substrate Reduction. J. Am. Chem. Soc. 1993, 115, 12193–12194. [Google Scholar] [CrossRef]
- Lovell, T.; Li, J.; Case, D.A.; Noodleman, L. FeMo cofactor of nitrogenase: Energetics and local interactions in the protein environment. J. Biol. Inorg. Chem. 2002, 7, 735–749. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, P.C.; Igarashi, R.Y.; Lee, H.I.; Hoffman, B.M.; Seefeldt, L.C.; Dean, D.R. Substrate interactions with the nitrogenase active site. Acc. Chem. Res. 2005, 38, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Lukoyanov, D.A.; Yang, Z.Y.; Perez-Gonzalez, A.; Raugei, S.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. 13C-ENDOR Characterization of the Central Carbon within the Nitrogenase Catalytic Cofactor Indicates That the CFe6 Core Is a Stabilizing “Heart of Steel”. J. Am. Chem. Soc. 2022, 2022, 6149. [Google Scholar]
- Lukoyanov, D.A.; Harris, D.F.; Yang, Z.Y.; Pérez-González, A.; Dean, D.R.; Seefeldt, L.C.; Hoffman, B.M. The One-Electron Reduced Active-Site FeFe-Cofactor of Fe-Nitrogenase Contains a Hydride Bound to a Formally Oxidized Metal-Ion Core. Inorg. Chem. 2022, 61, 5459–5464. [Google Scholar] [CrossRef]
- Bröcker, M.J.; Virus, S.; Ganskow, S.; Heathcote, P.; Heinz, D.W.; Schubert, W.D.; Jahn, D.; Moser, J. ATP-driven reduction by dark-operative protochlorophyllide oxidoreductase from Chlorobium tepidum mechanistically resembles nitrogenase catalysis. J. Biol. Chem. 2008, 283, 10559–10567. [Google Scholar] [CrossRef]
- Moore, S.J.; Sowa, S.T.; Schuchardt, C.; Deery, E.; Lawrence, A.D.; Ramos, J.V.; Billig, S.; Birkemeyer, C.; Chivers, P.T.; Howard, M.J.; et al. Elucidation of the biosynthesis of the methane catalyst coenzyme F430. Nature 2017, 543, 78–82. [Google Scholar] [CrossRef]
- Zheng, K.Y.; Ngo, P.D.; Owens, V.L.; Yang, X.P.; Mansoorabadi, S.O. The biosynthetic pathway of coenzyme F430 in methanogenic and methanotrophic archaea. Science 2016, 354, 339–342. [Google Scholar] [CrossRef]
- Garcia, A.K.; Harris, D.F.; Rivier, A.J.; Carruthers, B.M.; Pinochet-Barros, A.; Seefeldt, L.C.; Kacar, B. Nitrogenase resurrection and the evolution of a singular enzymatic mechanism. Elife 2023, 12, 1384. [Google Scholar] [CrossRef]
- Buckel, W.; Hetzel, M.; Kim, J. ATP-driven electron transfer in enzymatic radical reactions. Curr. Opin. Chem. Biol. 2004, 8, 462–467. [Google Scholar] [CrossRef]
- Seefeldt, L.C.; Peters, J.W.; Beratan, D.N.; Bothner, B.; Minteer, S.D.; Raugei, S.; Hoffman, B.M. Control of electron transfer in nitrogenase. Curr. Opin. Chem. Biol. 2018, 47, 54–59. [Google Scholar] [CrossRef]
- Rutledge, H.L.; Tezcan, F.A. Electron Transfer in Nitrogenase. Chem. Rev. 2020, 120, 5158–5193. [Google Scholar] [CrossRef] [PubMed]
- Burén, S.; Jimenez-Vicente, E.; Echavarri-Erasun, C.; Rubio, L.M. Biosynthesis of Nitrogenase Cofactors. Chem. Rev. 2020, 120, 4921–4968. [Google Scholar] [CrossRef]
- Wang, L.Y.; Zhang, L.H.; Liu, Z.Z.; Zhao, D.H.; Liu, X.M.; Zhang, B.; Xie, J.B.; Hong, Y.Y.; Li, P.F.; Chen, S.F.; et al. A Minimal Nitrogen Fixation Gene Cluster from Paenibacillus sp. WLY78 Enables Expression of Active Nitrogenase in Escherichia coli. PLoS Genet. 2013, 9, e1003865. [Google Scholar] [CrossRef]
- Yang, J.G.; Xie, X.Q.; Wang, X.; Dixon, R.; Wang, Y.P. Reconstruction and minimal gene requirements for the alternative iron-only nitrogenase in Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, E3718–E3725. [Google Scholar] [CrossRef] [PubMed]
- Wiig, J.A.; Lee, C.C.; Rebelein, J.G.; Sickerman, N.S.; Tanifuji, K.; Stiebritz, M.T.; Hu, Y.; Ribbe, M.W. Biosynthesis of the M-Cluster of Mo-Nitrogenase. Molybdenum Tungsten Enzym. Biochem. 2017, 5, 297–312. [Google Scholar]
- Dos Santos, P.C. Genomic manipulations of the diazotroph Azotobacter vinelandii. In Metalloproteins. Methods in Molecular Biology, 1st ed.; Hu, Y., Ed.; Humana Press: New York, NY, USA, 2019; Volume 1876, pp. 91–109. [Google Scholar]
- Brigle, K.E.; Setterquist, R.A.; Dean, D.R.; Cantwell, J.S.; Weiss, M.C.; Newton, W.E. Site-Directed Mutagenesis of the Nitrogenase MoFe Protein of Azotobacter vinelandii. Proc. Natl. Acad. Sci. USA 1987, 84, 7066–7069. [Google Scholar] [CrossRef]
- Hoffman, B.M.; Dean, D.R.; Seefeldt, L.C. Climbing Nitrogenase: Toward a Mechanism of Enzymatic Nitrogen Fixation. Acc. Chem. Res. 2009, 42, 609–619. [Google Scholar] [CrossRef]
- Smil, V. Detonator of the population explosion. Nature 1999, 400, 415. [Google Scholar] [CrossRef]
- Erisman, J.W.; Sutton, M.A.; Galloway, J.; Klimont, Z.; Winiwarter, W. How a century of ammonia synthesis changed the world. Nat. Geosci. 2008, 1, 636–639. [Google Scholar] [CrossRef]
- Gu, B.J.; Zhang, X.M.; Lam, S.K.; Yu, Y.L.; van Grinsven, H.J.M.; Zhang, S.H.; Wang, X.X.; Bodirsky, B.L.; Wang, S.T.; Duan, J.K.; et al. Cost-effective mitigation of nitrogen pollution from global croplands. Nature 2023, 613, 77–84. [Google Scholar] [CrossRef]
- Buren, S.; Rubio, L.M. State of the art in eukaryotic nitrogenase engineering. FEMS Microbiol. Lett. 2018, 365, 2. [Google Scholar] [CrossRef] [PubMed]
- Johnston, E.; Okada, S.; Gregg, C.M.; Warden, A.C.; Rolland, V.; Gillespie, V.; Byrne, K.; Colgrave, M.L.; Eamens, A.L.; Allen, R.S.; et al. The structural components of the iron-only nitrogenase, AnfDKG, form a protein complex within the plant mitochondrial matrix. Plant Mol. Biol. 2023, 112, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Gregg, C.M.; Allen, R.S.; Menon, A.; Hussain, D.; Gillespie, V.; Johnston, E.; Byrne, K.; Colgrave, M.L.; Wood, C.C. A Synthetic Biology Workflow Reveals Variation in Processing and Solubility of Nitrogenase Proteins Targeted to Plant Mitochondria, and Differing Tolerance of Targeting Sequences in a Bacterial Nitrogenase Assay. Front. Plant Sci. 2020, 11, 552160. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Components of the nitrogenase system and their interaction. A low-potential ferredoxin of flavodoxin reduces the reductase component (or Fe protein), which then binds 2 ATP and undergoes a conformational change that allows for complex formation with the dinitrogenase component. This triggers ATP hydrolysis, upon which an electron is transferred from the [8Fe:7S] P-cluster to the active-site cofactor, a [Mo:7Fe:9S:C]:homocitrate cluster in the case of Mo-nitrogenase (red arrows). The reduction of a single N2 molecule to 2 NH4+ requires this cycle to be repeated eight times at the expense of 16 ATP.
Figure 1.
Components of the nitrogenase system and their interaction. A low-potential ferredoxin of flavodoxin reduces the reductase component (or Fe protein), which then binds 2 ATP and undergoes a conformational change that allows for complex formation with the dinitrogenase component. This triggers ATP hydrolysis, upon which an electron is transferred from the [8Fe:7S] P-cluster to the active-site cofactor, a [Mo:7Fe:9S:C]:homocitrate cluster in the case of Mo-nitrogenase (red arrows). The reduction of a single N2 molecule to 2 NH4+ requires this cycle to be repeated eight times at the expense of 16 ATP.

Figure 3.
The kinetic scheme for nitrogenase catalysis according to Lowe and Thorneley [37]. The 8-electron process that reduces N2 to 2 NH4+ and releases a stoichiometric H2 molecule upon substrate binding is defined in eight states, E0–E7, that correspond to single-electron transfer events. The enzyme must reach the E4 state to bind N2 in exchange for H2, which is interpreted as a reductive elimination that leaves the enzyme in a super-reduced state.
Figure 3.
The kinetic scheme for nitrogenase catalysis according to Lowe and Thorneley [37]. The 8-electron process that reduces N2 to 2 NH4+ and releases a stoichiometric H2 molecule upon substrate binding is defined in eight states, E0–E7, that correspond to single-electron transfer events. The enzyme must reach the E4 state to bind N2 in exchange for H2, which is interpreted as a reductive elimination that leaves the enzyme in a super-reduced state.

Figure 4.
The three variants of nitrogenase. (a) Mo-nitrogenase is encoded by nif genes and consists of the Fe protein NifH2 and the MoFe protein NifD2K2 (above). MoFe protein contains [8Fe:7S] P-clusters at the DK interfaces (middle) and an active-site FeMo cofactor (below). This cofactor is a pseudo-D32 symmetric [Mo:7Fe:9S:C]:homocitrate cluster attached to the protein only through its apical metal ions. (b) In the alternative V-nitrogenase, encoded by vnf genes, the Fe protein VnfH2 works in conjunction with the VFe protein VnfD2K2G2 (above). The P-cluster corresponds fully to that of the MoFe protein (middle), and the active-site FeV cofactor had Mo replaced for V, as expected (below). What was unexpected was the replacement of one µ2-sulfide, S3A, by a carbonate anion that is not exchanged during catalysis. (c) In Fe-nitrogenase, anf genes encode the Fe protein AnfH2 and the FeFe protein Anf D2K2G2 (above). Although FeFe and VFe proteins are closely related, their Fe proteins AnfH and VnfH are more distinct than VnfH and NifH. The P-cluster of FeFe protein is highly similar to those of the isoenzymes (middle), and the FeFe cofactor (below) is a truly D32 symmetric [8Fe:9S:C]:homocitrate cluster that likely corresponds to a precursor (L-cluster) of the other sites.
Figure 4.
The three variants of nitrogenase. (a) Mo-nitrogenase is encoded by nif genes and consists of the Fe protein NifH2 and the MoFe protein NifD2K2 (above). MoFe protein contains [8Fe:7S] P-clusters at the DK interfaces (middle) and an active-site FeMo cofactor (below). This cofactor is a pseudo-D32 symmetric [Mo:7Fe:9S:C]:homocitrate cluster attached to the protein only through its apical metal ions. (b) In the alternative V-nitrogenase, encoded by vnf genes, the Fe protein VnfH2 works in conjunction with the VFe protein VnfD2K2G2 (above). The P-cluster corresponds fully to that of the MoFe protein (middle), and the active-site FeV cofactor had Mo replaced for V, as expected (below). What was unexpected was the replacement of one µ2-sulfide, S3A, by a carbonate anion that is not exchanged during catalysis. (c) In Fe-nitrogenase, anf genes encode the Fe protein AnfH2 and the FeFe protein Anf D2K2G2 (above). Although FeFe and VFe proteins are closely related, their Fe proteins AnfH and VnfH are more distinct than VnfH and NifH. The P-cluster of FeFe protein is highly similar to those of the isoenzymes (middle), and the FeFe cofactor (below) is a truly D32 symmetric [8Fe:9S:C]:homocitrate cluster that likely corresponds to a precursor (L-cluster) of the other sites.

Figure 5.
CO inhibition of the nitrogenase cofactor. Using the FeV cofactor as an example, the resting state is reduced by two electrons, leading to the release of sulfide S2B and its replacement by a bridging hydride in the E2 state. Here, CO can bind terminally to the t-site at Fe6. If the µ-hydride is accidentally protonated and lost as H2, the CO ligand can migrate to the µ-site, but the enzyme is formally returned to the E0 state, now in a CO-inhibited form. Pressurization of crystals of this low-CO state with CO gas led to the structure of the high-CO state that still is an off-pathway resting state E0, but with two CO ligands that highlight the µ- and t-site for substrate/intermediate binding.
Figure 5.
CO inhibition of the nitrogenase cofactor. Using the FeV cofactor as an example, the resting state is reduced by two electrons, leading to the release of sulfide S2B and its replacement by a bridging hydride in the E2 state. Here, CO can bind terminally to the t-site at Fe6. If the µ-hydride is accidentally protonated and lost as H2, the CO ligand can migrate to the µ-site, but the enzyme is formally returned to the E0 state, now in a CO-inhibited form. Pressurization of crystals of this low-CO state with CO gas led to the structure of the high-CO state that still is an off-pathway resting state E0, but with two CO ligands that highlight the µ- and t-site for substrate/intermediate binding.

Figure 6.
Proposed mechanistic framework for nitrogenase on the example of CO reduction to the methyl stage by V- and Fe-nitrogenase. Two-electron reduction from the resting E0 state leads to the formation of a terminal hydride at the t-site, Fe6. This t-H inserts into the bound ligand, and the cycle is repeated, leading through formyl and hydroxymethyl adducts to C-O bond cleavage, water release, and a methyl group bound to the enzyme.
Figure 6.
Proposed mechanistic framework for nitrogenase on the example of CO reduction to the methyl stage by V- and Fe-nitrogenase. Two-electron reduction from the resting E0 state leads to the formation of a terminal hydride at the t-site, Fe6. This t-H inserts into the bound ligand, and the cycle is repeated, leading through formyl and hydroxymethyl adducts to C-O bond cleavage, water release, and a methyl group bound to the enzyme.

Figure 7.
Proposed electronic structure of the resting state E0 for the nitrogenase cofactors. Only metals are shown. Sulfides and the central carbide are omitted for clarity. The arrow in each metal indicates the spin orientation for the high-spin systems, following the BS7 coupling scheme. As a common principle, Fe2 and Fe6 emerge as the most oxidized sites, and Fe6 takes up the electron from the Fe protein via the P-cluster. In all clusters, three additional electrons are distributed across Fe1, Fe3, Fe4, Fe5, and Fe7, with delocalization between Fe3–4 and Fe5–7. Only the heterometals or Fe8 in the FeFe cofactor have an octahedral ligand field. In this proposal, the resting state configurations add up to the apparent spins of S = 3/2 for FeMo cofactor and integer spins for FeV and FeFe cofactors.
Figure 7.
Proposed electronic structure of the resting state E0 for the nitrogenase cofactors. Only metals are shown. Sulfides and the central carbide are omitted for clarity. The arrow in each metal indicates the spin orientation for the high-spin systems, following the BS7 coupling scheme. As a common principle, Fe2 and Fe6 emerge as the most oxidized sites, and Fe6 takes up the electron from the Fe protein via the P-cluster. In all clusters, three additional electrons are distributed across Fe1, Fe3, Fe4, Fe5, and Fe7, with delocalization between Fe3–4 and Fe5–7. Only the heterometals or Fe8 in the FeFe cofactor have an octahedral ligand field. In this proposal, the resting state configurations add up to the apparent spins of S = 3/2 for FeMo cofactor and integer spins for FeV and FeFe cofactors.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Einsle, O. On the Shoulders of Giants—Reaching for Nitrogenase. Molecules 2023, 28, 7959. https://doi.org/10.3390/molecules28247959
AMA Style
Einsle O. On the Shoulders of Giants—Reaching for Nitrogenase. Molecules. 2023; 28(24):7959. https://doi.org/10.3390/molecules28247959
Chicago/Turabian StyleEinsle, Oliver. 2023. "On the Shoulders of Giants—Reaching for Nitrogenase" Molecules 28, no. 24: 7959. https://doi.org/10.3390/molecules28247959