
Schisandrin B Alleviates Renal Tubular Cell Epithelial–Mesenchymal Transition and Mitochondrial Dysfunction by Kielin/Chordin-like Protein Upregulation via Akt Pathway Inactivation and Adenosine 5′-Monophosphate (AMP)-Activated Protein Kinase Pathway Activation in Diabetic Kidney Disease
Abstract
:1. Introduction
2. Results
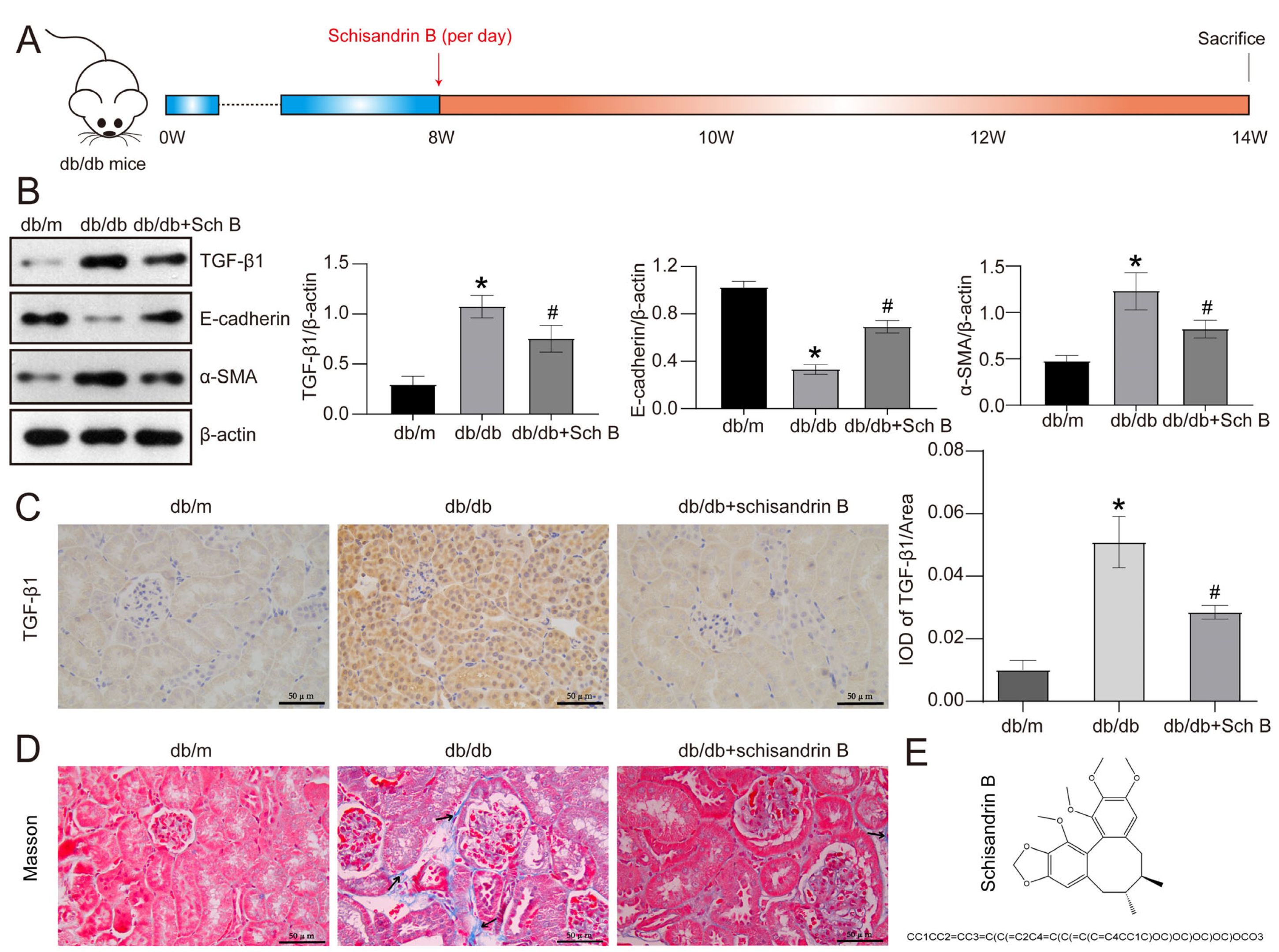
2.1. Schisandrin B Attenuates Epithelial–Mesenchymal Transition of Renal Tubular Cells in db/db Mice
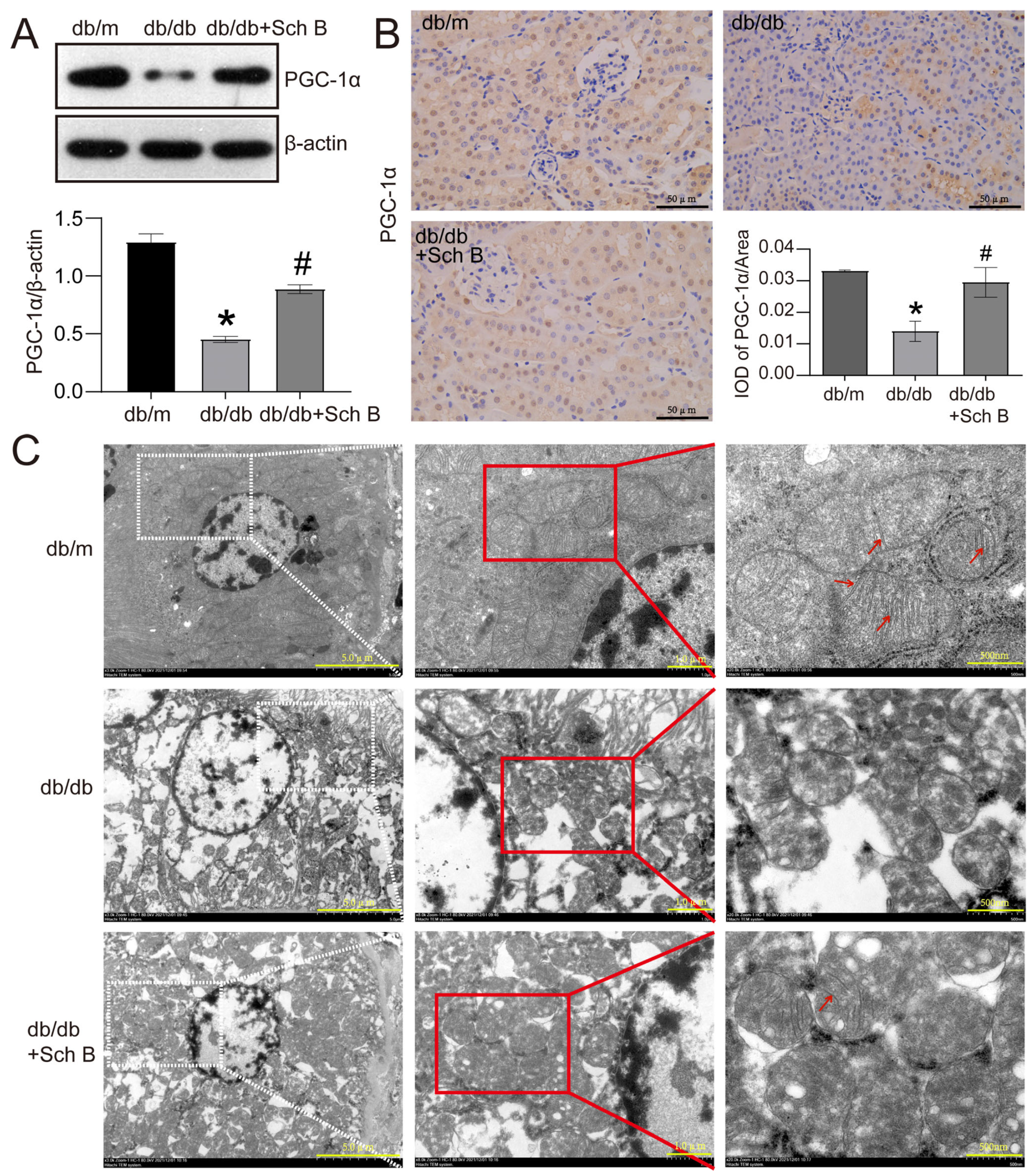
2.2. Schisandrin B Alleviates Mitochondrial Dysfunction of Renal Tubular Cells from db/db Mice
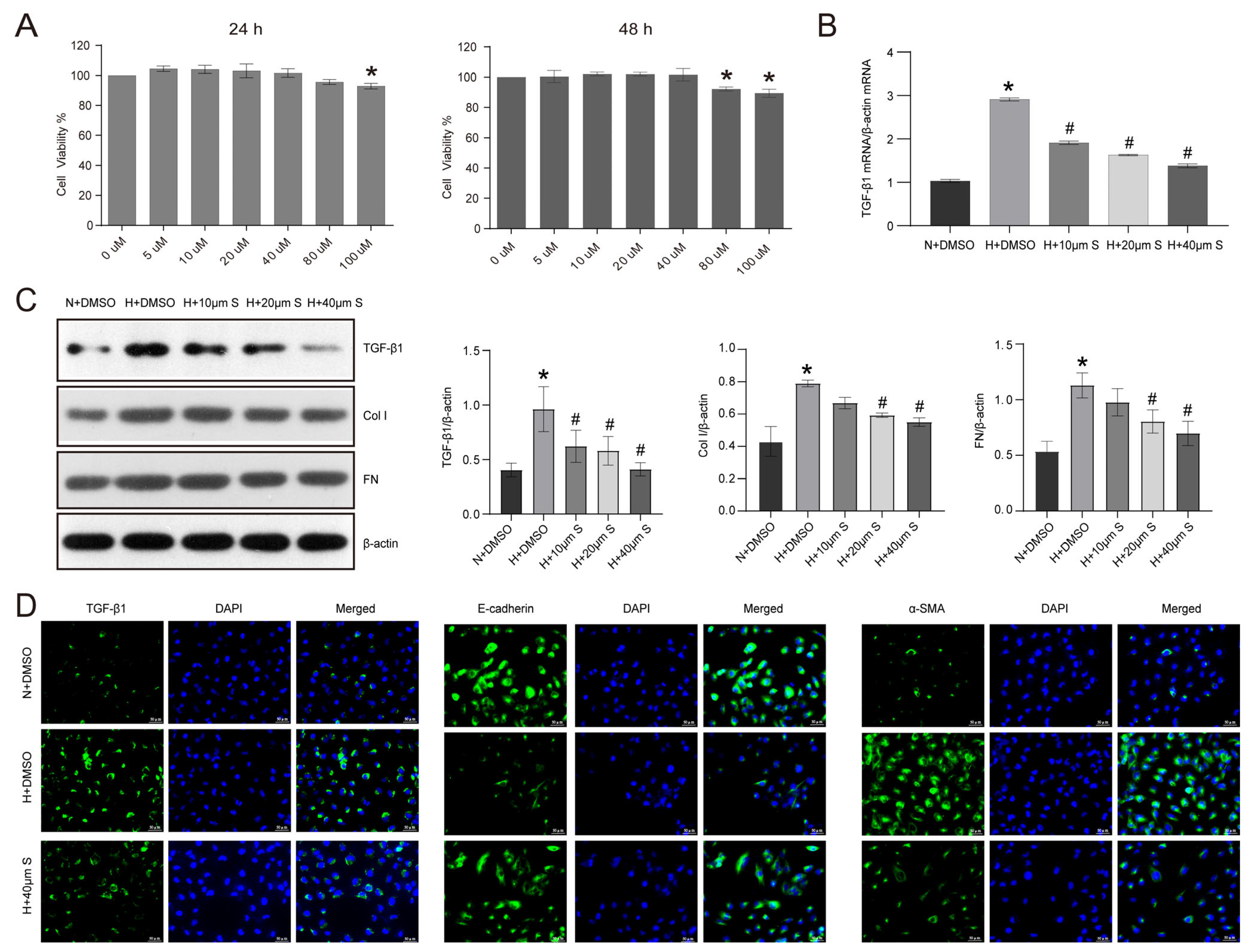
2.3. Schisandrin B Inhibits Epithelial–Mesenchymal Transition in HK2 Cells Exposed to Elevated Levels of Glucose
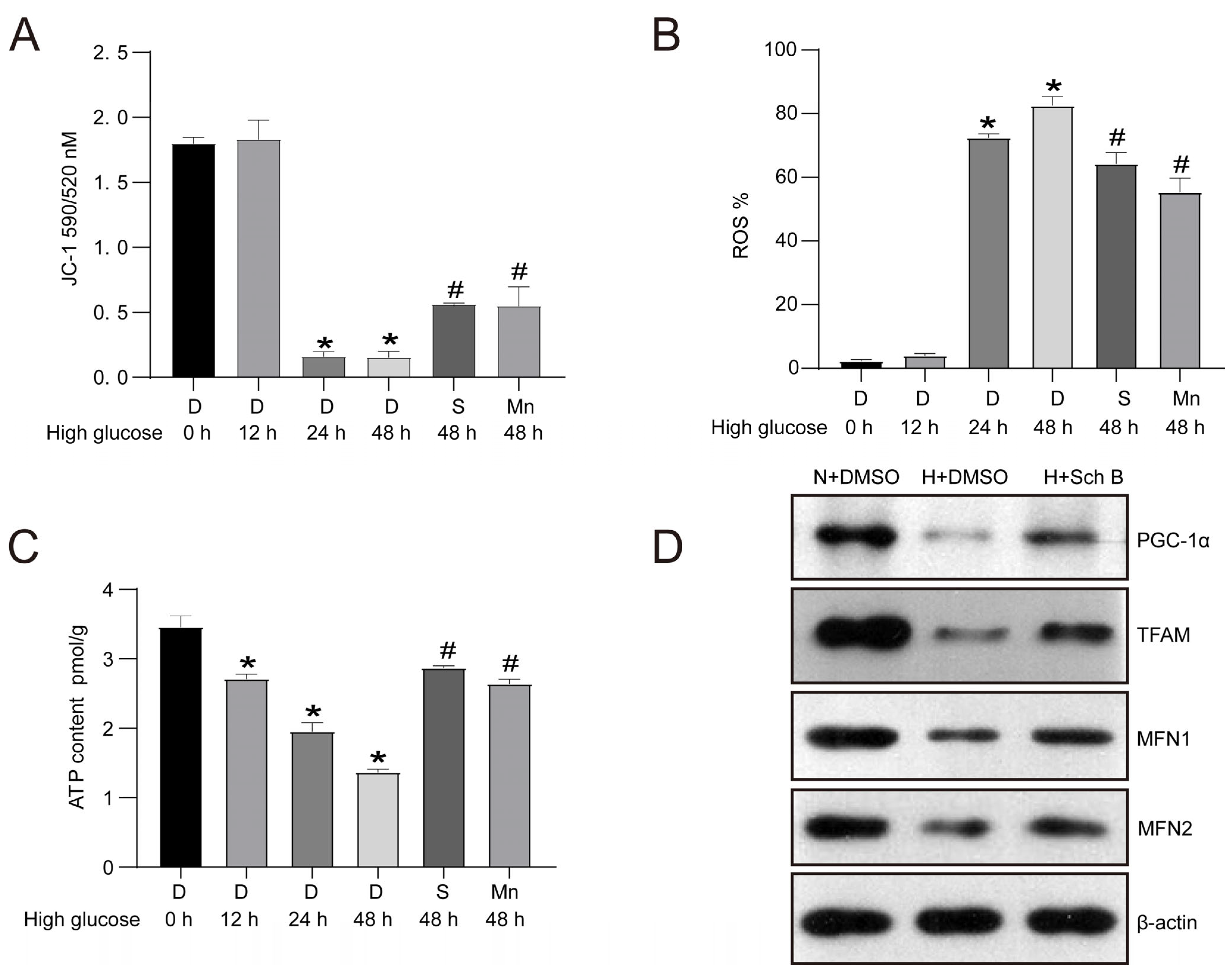
2.4. Schisandrin B Improves Mitochondrial Dysfunction in HK2 Cells Exposed to Elevated Levels of Glucose
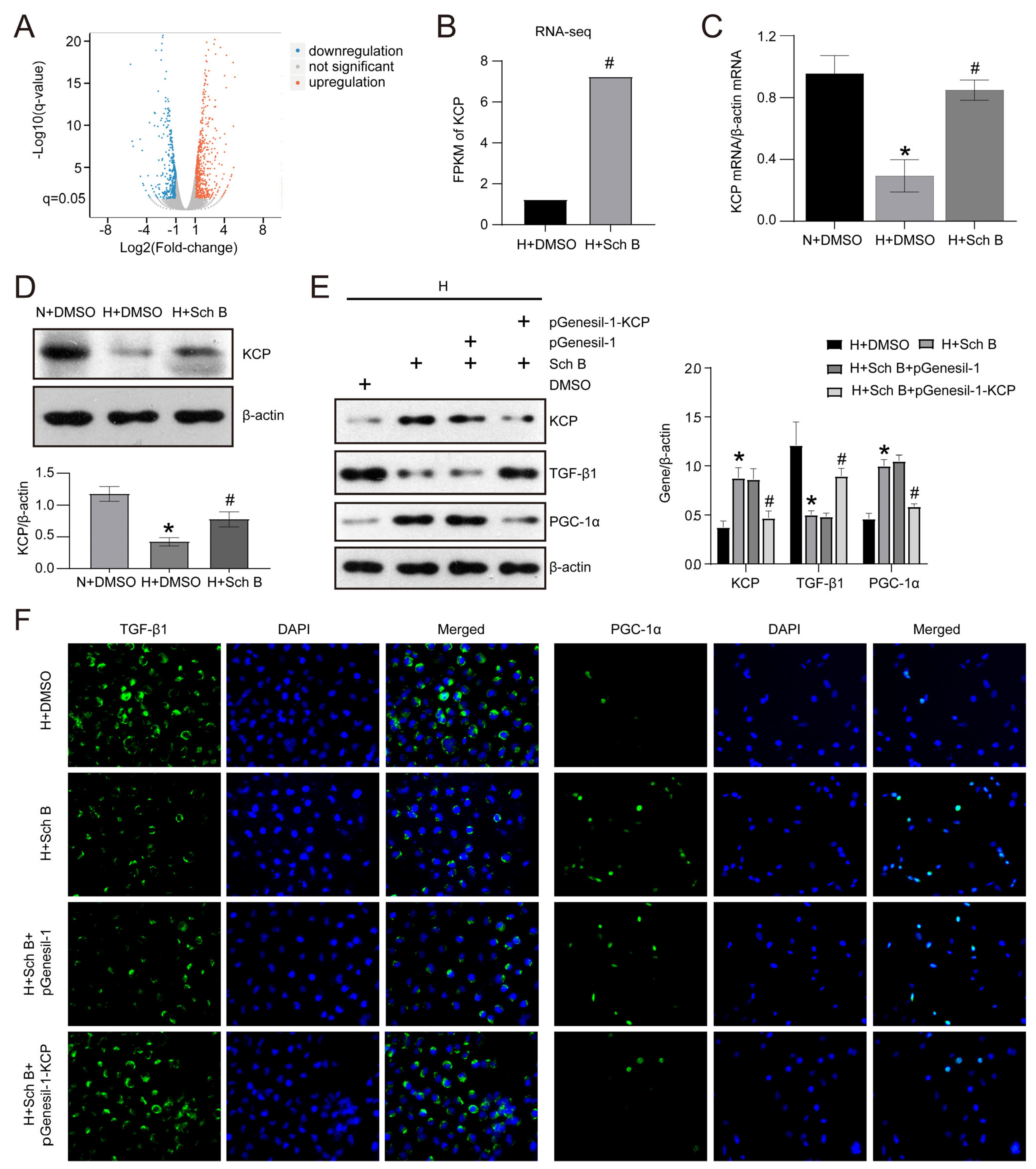
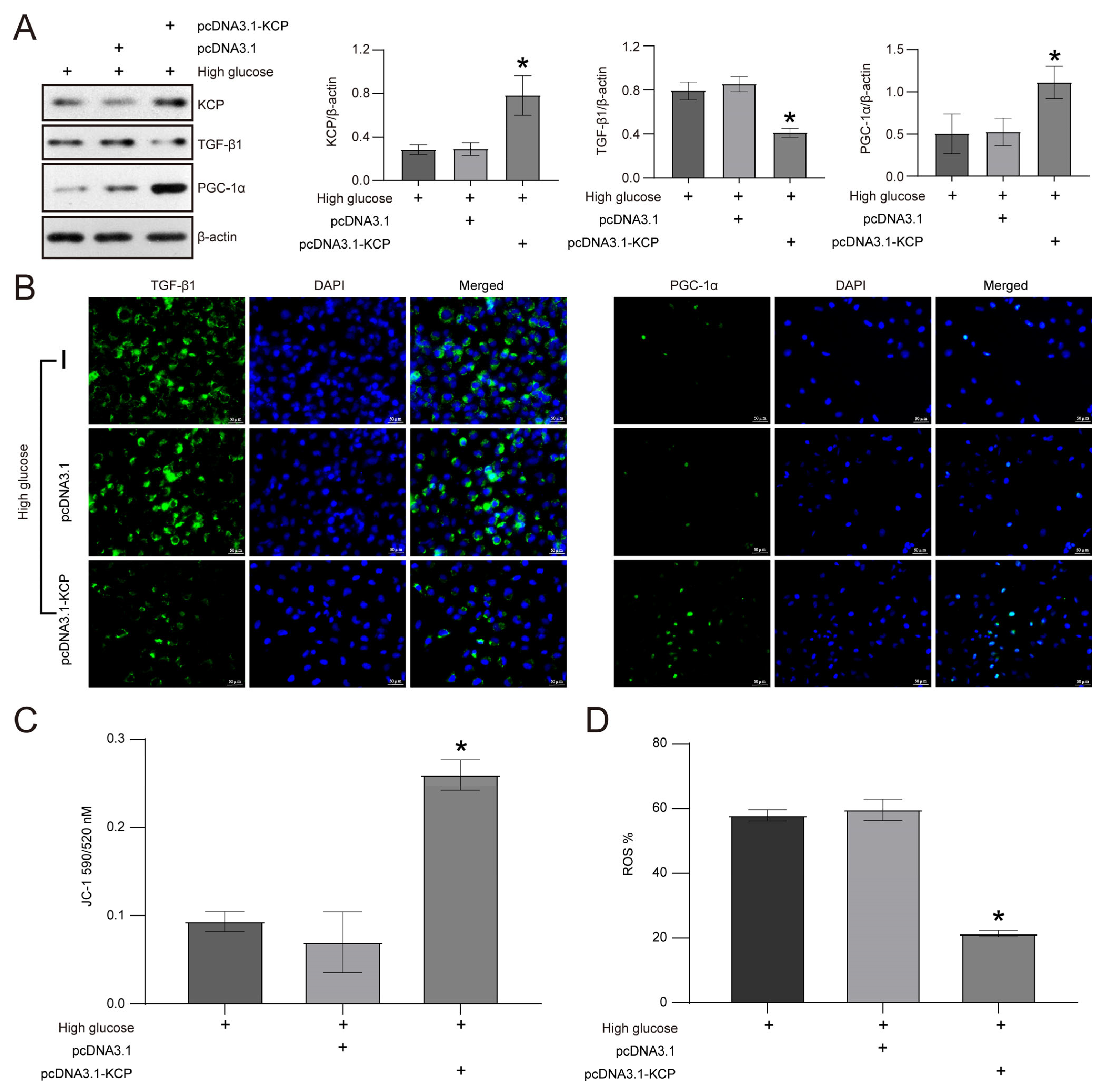
2.5. KCP Upregulation Mediates Schisandrin B-Regulated Epithelial–Mesenchymal Transition and Mitochondrial Dysfunction in HK2 Cells Exposed to High Glucose Levels
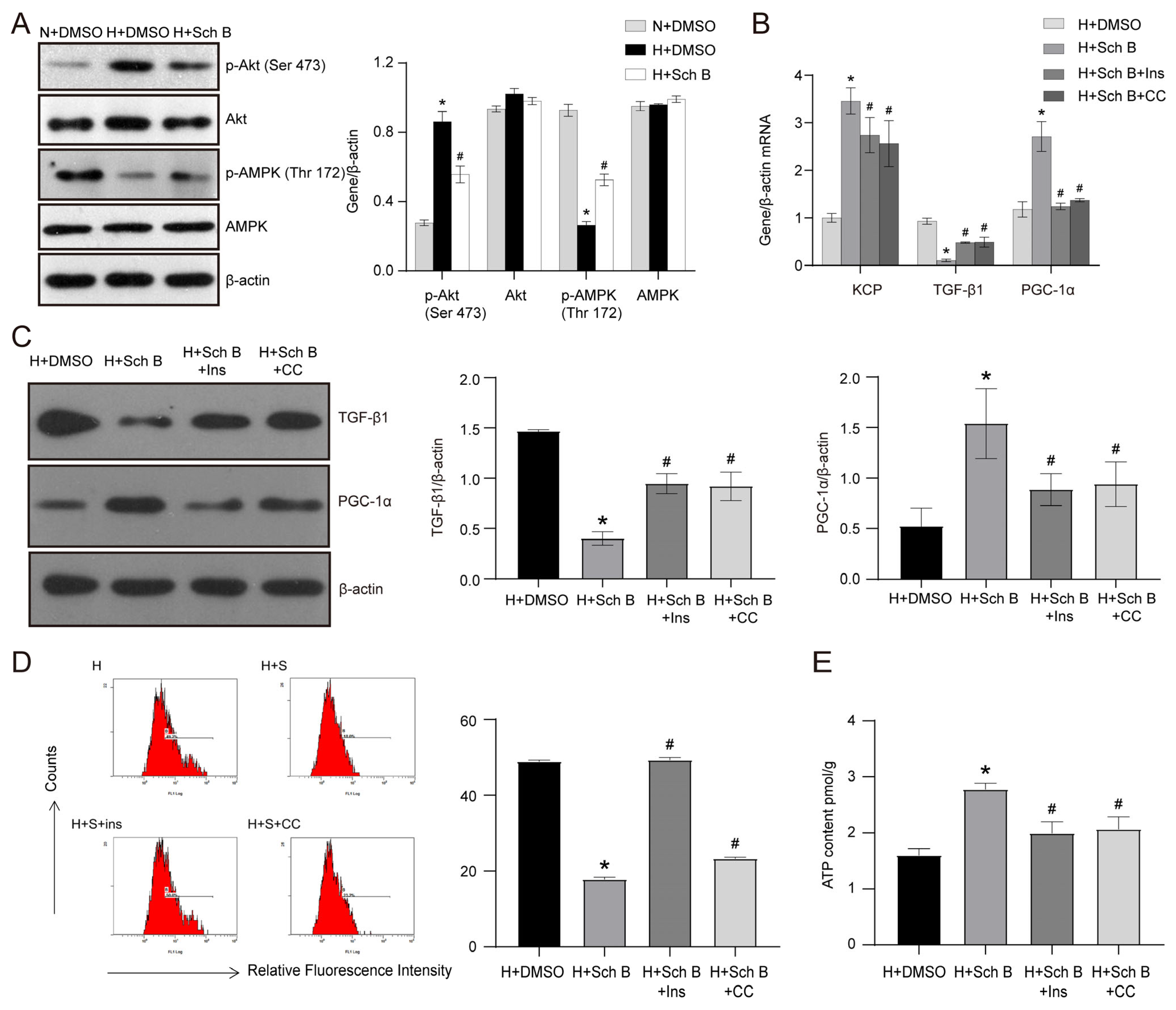
2.6. PI3K/Akt Pathway Inhibition and AMPK Pathway Activation Are Involved in Schisandrin B-Induced Increase in KCP Expression in HK2 Cells Exposed to High Glucose Levels
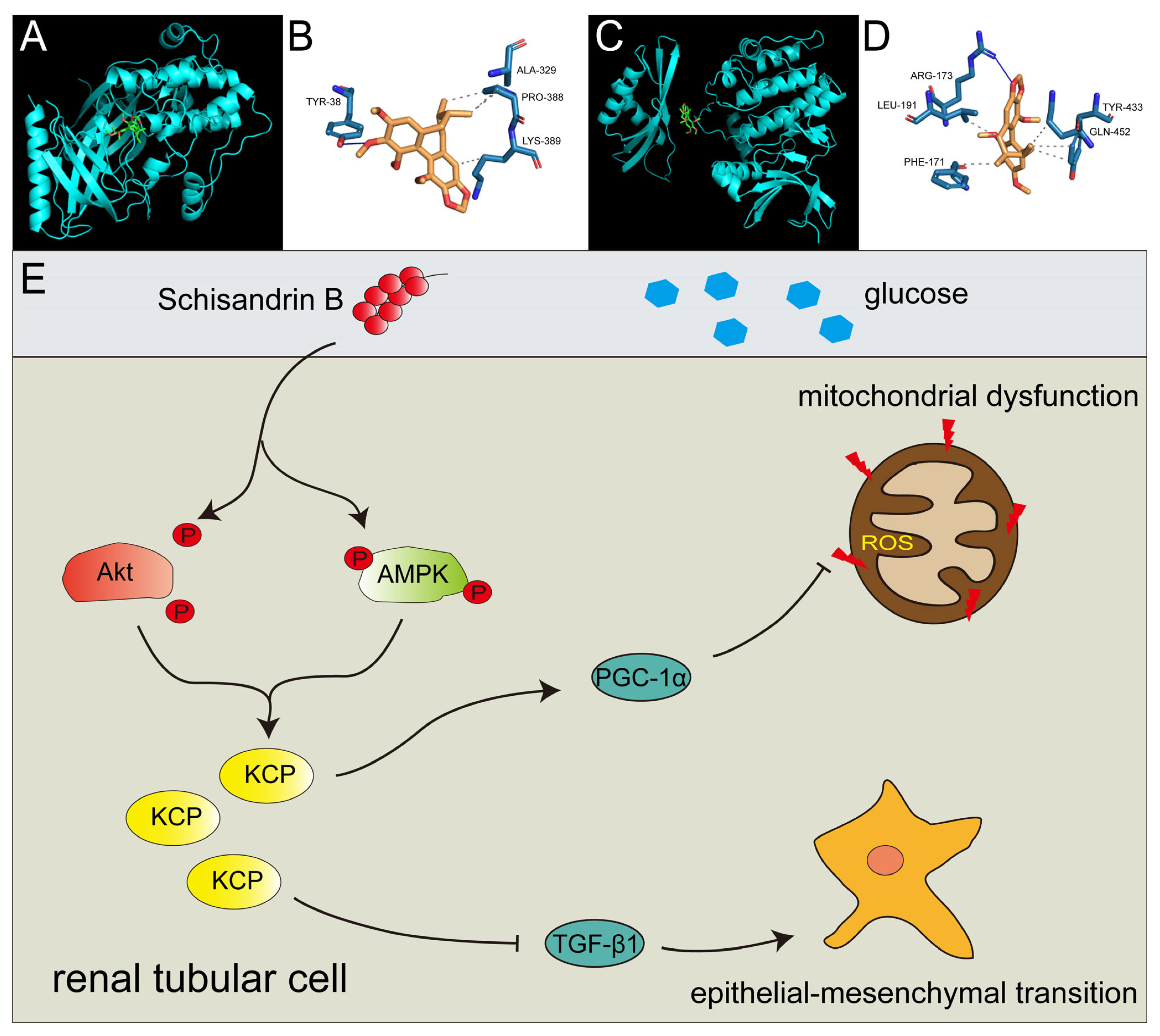
2.7. Schisandrin B Could Bind with Akt at Ser 473 Phosphorylation Site and AMPK at Thr 172 Phosphorylation Site
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animal Experiment
4.3. Cell Culture
4.4. Cell Transfection
4.5. Western Blotting
4.6. Immunohistochemistry
4.7. Immunofluorescence
4.8. Intracellular ROS Assay
4.9. ATP Content Assay
4.10. JC-1-Mitochondrial Membrane Potential Assay
4.11. Cell Viability Detection
4.12. Electron Microscopy
4.13. RNA-Seq
4.14. Real-Time PCR
4.15. Molecular Autodock Analysis
4.16. Masson Trichrome Staining
4.17. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Akhtar, M.; Taha, N.M.; Nauman, A.; Mujeeb, I.B.; Al-Nabet, A.D.M. Diabetic Kidney Disease: Past and Present. Adv. Anat. Pathol. 2020, 27, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Reidy, K.; Kang, H.M.; Hostetter, T.; Susztak, K. Molecular mechanisms of diabetic kidney disease. J. Clin. Investig. 2014, 124, 2333–2340. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Livingston, M.J.; Liu, Z.; Dong, G.; Zhang, M.; Chen, J.-K.; Dong, Z. Autophagy in diabetic kidney disease: Regulation, pathological role and therapeutic potential. Cell. Mol. Life Sci. 2017, 75, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Luo, Y.; Yang, S.; Zeng, M.; Zhang, S.; Liu, J.; Han, Y.; Liu, Y.; Zhu, X.; Wu, H.; et al. Ectopic lipid accumulation: Potential role in tubular injury and inflammation in diabetic kidney disease. Clin. Sci. 2018, 132, 2407–2422. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lv, L.; Liu, B.; Tang, R. Crosstalk between tubular epithelial cells and glomerular endothelial cells in diabetic kidney disease. Cell Prolif. 2020, 53, e12763. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.; Hsu, Y.; Shih, Y.; Chang, P.; Lin, C. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology 2018, 23 (Suppl. S4), 32–37. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhao, S.; Liu, S.; Liu, Q.; Li, F.; Hao, J. PTEN Regulates Renal Extracellular Matrix Deposit via Increased CTGF in Diabetes Mellitus. J. Cell. Biochem. 2016, 117, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Luo, J.; Zhao, Z.; Liao, Y.; Li, Y. Tranilast prevents renal interstitial fibrosis by blocking mast cell infiltration in a rat model of diabetic kidney disease. Mol. Med. Rep. 2018, 17, 7356–7364. [Google Scholar] [CrossRef]
- Jun, H.; Song, Z.; Chen, W.; Zanhua, R.; Yonghong, S.; Shuxia, L.; Huijun, D. In vivo and in vitro effects of SREBP-1 on diabetic renal tubular lipid accumulation and RNAi-mediated gene silencing study. Histochem. 2009, 131, 327–345. [Google Scholar] [CrossRef]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Johri, A.; Chandra, A.; Beal, M.F. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Mise, K.; Danesh, F.R. Mitochondrial Regulation of Diabetic Kidney Disease. Front. Med. 2021, 8, 745279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chi, Y.; Kang, Y.; Lu, H.; Niu, H.; Liu, W.; Li, Y. Resveratrol ameliorates podocyte damage in diabetic mice via SIRT1/PGC-1α mediated attenuation of mitochondrial oxidative stress. J. Cell. Physiol. 2018, 234, 5033–5043. [Google Scholar] [CrossRef] [PubMed]
- Galvan, D.L.; Green, N.H.; Danesh, F.R. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int. 2017, 92, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.I.; Zhu, S.; Chen, C.; Zhao, M.; Huang, H.; Zhu, P. A Comprehensive Review on Schisandrin B and Its Biological Properties. Oxid. Med. Cell. Longev. 2020, 2020, 2172740. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Yan, W.; Cui, X.; Ma, W.; Wang, Z.; Liu, N.; Yi, X.; Guo, T.; Wei, X.; Sun, Y.; et al. Schisandrin B, a potential GLP-1R agonist, exerts anti-diabetic effects by stimulating insulin secretion. Mol. Cell. Endocrinol. 2023, 577, 112029. [Google Scholar] [CrossRef]
- Song, J.; Zhang, B.; Zhang, H.; Cheng, W.; Liu, P.; Kang, J. Quantitative proteomics combined with network pharmacology analysis unveils the biological basis of Schisandrin B in treating diabetic nephropathy. Comb. Chem. High Throughput Screen. 2023, 17. [Google Scholar] [CrossRef]
- Mou, Z.; Feng, Z.; Xu, Z.; Zhuang, F.; Zheng, X.; Li, X.; Qian, J.; Liang, G. Schisandrin B alleviates diabetic nephropathy through suppressing excessive inflammation and oxidative stress. Biochem. Biophys. Res. Commun. 2019, 243–249. [Google Scholar] [CrossRef]
- Yanagita, M. Modulator of bone morphogenetic protein activity in the progression of kidney diseases. Kidney Int. 2006, 70, 989–993. [Google Scholar] [CrossRef]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Cheng, Y.; Han, F.; Chang, Y.; Yang, Y.; Li, X.; Chen, L.; Lu, Y.; Sun, B.; Chen, L. Triptolide Attenuates Renal Tubular Epithelial-mesenchymal Transition Via the MiR-188-5p-mediated PI3K/AKT Pathway in Diabetic Kidney Disease. Int. J. Biol. Sci. 2018, 14, 1545–1557. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Yang, F.; Le, Y.; Yang, Y.; Wang, B.; Jia, Y.; Zheng, Z.; Xue, Y. Klotho protects against diabetic kidney disease via AMPK- and ERK-mediated autophagy. Acta Diabetol. 2021, 58, 1413–1423. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wang, H.; Ma, R.; Wang, L. Schisandrin B inhibits epithelial transition and stemness of large lung cancer cells and tumorigenesis in xenografts via inhibiting the NF and p38 MAPK signaling pathways. Oncol. Rep. 2021, 45, 115. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, B.; Liu, K.; Ding, Z.; Hu, X. Schisandrin B Attenuates Cancer Invasion and Metastasis Via Inhibiting Epithelial-Mesenchymal Transition. PLoS ONE 2012, 7, e40480. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Li, S.; Shi, H.; Yin, P.; Chen, J.; Li, H.; Zhong, Y.; Diao, L.-T.; Du, B. Schisandrin B attenuates renal fibrosis via miR-30e-mediated inhibition of EMT. Toxicol. Appl. Pharmacol. 2019, 385, 114769. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Shi, X.; Zheng, Z.; Zhang, B.; Shi, F.; Jiang, L.; Xu, J. Schisandrin B protects against angiotensin II-induced endotheliocyte deficits by targeting Keap1 and activating Nrf2 pathway. Drug Des. Dev. Ther. 2018, 12, 3985–3997. [Google Scholar] [CrossRef]
- Guo, M.; An, F.; Yu, H.; Wei, X.; Hong, M.; Lu, Y. Comparative effects of schisandrin A, B, and C on Propionibacterium acnes-induced, NLRP3 inflammasome activation-mediated IL-1β secretion and pyroptosis. Biomed. Pharmacother. 2017, 96, 129–136. [Google Scholar] [CrossRef]
- Wang, J.; Fang, Z.; Song, C.; Kang, H.; Guo, Q.; Dong, Y.; Zhang, Y.; Peng, R.; Guan, H.; Li, F. Schisandrin B Inhibits Osteoclastogenesis and Protects Against Ovariectomy-Induced Bone Loss. Front. Pharmacol. 2020, 11, 1175. [Google Scholar] [CrossRef]
- Soofi, A.; Zhang, P.; Dressler, G.R. Kielin/Chordin-Like Protein Attenuates both Acute and Chronic Renal Injury. J. Am. Soc. Nephrol. 2013, 24, 897–905. [Google Scholar] [CrossRef]
- Soofi, A.; Wolf, K.I.; Emont, M.P.; Qi, N.; Martinez-Santibanez, G.; Grimley, E.; Ostwani, W.; Dressler, G.R. The kielin/chordin-like protein (KCP) attenuates high-fat diet-induced obesity and metabolic syndrome in mice. J. Biol. Chem. 2017, 292, 9051–9062. [Google Scholar] [CrossRef] [PubMed]
- Bhattamisra, S.K.; Koh, H.M.; Lim, S.Y.; Choudhury, H.; Pandey, M. Molecular and Biochemical Pathways of Catalpol in Alleviating Diabetes Mellitus and Its Complications. Biomolecules 2021, 11, 323. [Google Scholar] [CrossRef]
- Ravindran, S.; Kuruvilla, V.; Wilbur, K.; Munusamy, S. Nephroprotective Effects of Metformin in Diabetic Nephropathy. J. Cell. Physiol. 2017, 232, 731–742. [Google Scholar] [CrossRef]
- Wang, B.; Wang, X.; Tong, X.; Zhang, Y. Schisandrin B Inhibits Cell Viability and Migration, and Induces Cell Apoptosis by circ_0009112/miR-708-5p Axis Through PI3K/AKT Pathway in Osteosarcoma. Front. Genet. 2020, 11, 588670. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, G.-P.; Peng, J.-J.; Ren, L.-H.; Lei, L.-C.; Ye, H.-M.; Wang, Z.-Y.; Zhao, S. Schizandrin B attenuates hypoxia/reoxygenation injury in H9c2 cells by activating the AMPK/Nrf2 signaling pathway. Exp. Ther. Med. 2021, 21, 220. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | db/m | db/db | db/db + Sch B |
|---|---|---|---|
| Body weight (g) | 25.60 ± 0.55 | 55.60 ± 1.66 * | 50.24 ± 4.06 |
| Food intake (g/day) | 3.81 ± 0.24 | 9.96 ± 0.22 * | 9.35 ± 0.46 |
| Daily urinary volume (mL) | 0.33 ± 0.13 | 4.10 ± 0.48 * | 4.15 ± 0.84 |
| Kidney weight (g) | 0.14 ± 0.008 | 0.18 ± 0.011 * | 0.19 ± 0.016 |
| Kidney weight/body weight (%) | 0.56 ± 0.032 | 0.32 ± 0.027 * | 0.38 ± 0.054 |
| Blood glucose (mmol/L) | 6.25 ± 0.42 | 29.43 ± 2.17 * | 23.48 ± 5.74 |
| Serum creatinine (μmol/L) | 8.68 ± 0.33 | 17.33 ± 4.92 * | 8.73 ± 0.83 # |
| Blood urea nitrogen (mmol/L) | 5.90 ± 0.35 | 8.35 ± 0.29 * | 7.41 ± 0.67 |
| Cystatin C (mg/L) | 0.28 ± 0.05 | 0.34 ± 0.04 | 0.28 ± 0.10 |
| Urine albumin excretion (μg/24 h) | 17.0 ± 6.85 | 516.76 ± 60.91 * | 237.68 ± 49.29 # |
| Forward Primer | Reverse Primer | Product | |
|---|---|---|---|
| Human KCP | GGGACACCAGTATCAGAGCCA | CCCCATCTTGACAGACGCAG | 245 bp |
| Human TGF-β1 | AGCAACAATTCCTGGCGATAC | CTAAGGCGAAAGCCCTCAAT | 137 bp |
| Human PGC-1α | ACACTTTGCGCAGGTCAAAC | AGCAGGGTCAAAGTCATCTGAG | 116 bp |
| Human β-actin | AAGGCCAACCGCGAGAA | ATGGGGGAGGGCATACC | 183 bp |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, W.; Li, F.; Guo, D.; Du, C.; Zhao, S.; Li, J.; Yan, Z.; Hao, J. Schisandrin B Alleviates Renal Tubular Cell Epithelial–Mesenchymal Transition and Mitochondrial Dysfunction by Kielin/Chordin-like Protein Upregulation via Akt Pathway Inactivation and Adenosine 5′-Monophosphate (AMP)-Activated Protein Kinase Pathway Activation in Diabetic Kidney Disease. Molecules 2023, 28, 7851. https://doi.org/10.3390/molecules28237851
Liu W, Li F, Guo D, Du C, Zhao S, Li J, Yan Z, Hao J. Schisandrin B Alleviates Renal Tubular Cell Epithelial–Mesenchymal Transition and Mitochondrial Dysfunction by Kielin/Chordin-like Protein Upregulation via Akt Pathway Inactivation and Adenosine 5′-Monophosphate (AMP)-Activated Protein Kinase Pathway Activation in Diabetic Kidney Disease. Molecules. 2023; 28(23):7851. https://doi.org/10.3390/molecules28237851
Chicago/Turabian StyleLiu, Weilin, Fan Li, Dongwei Guo, Congyuan Du, Song Zhao, Juan Li, Zhe Yan, and Jun Hao. 2023. "Schisandrin B Alleviates Renal Tubular Cell Epithelial–Mesenchymal Transition and Mitochondrial Dysfunction by Kielin/Chordin-like Protein Upregulation via Akt Pathway Inactivation and Adenosine 5′-Monophosphate (AMP)-Activated Protein Kinase Pathway Activation in Diabetic Kidney Disease" Molecules 28, no. 23: 7851. https://doi.org/10.3390/molecules28237851