
Metal-Free One-Pot Multi-Functionalization of Unsaturated Compounds with Interelement Compounds by Radical Process

{kind=link}
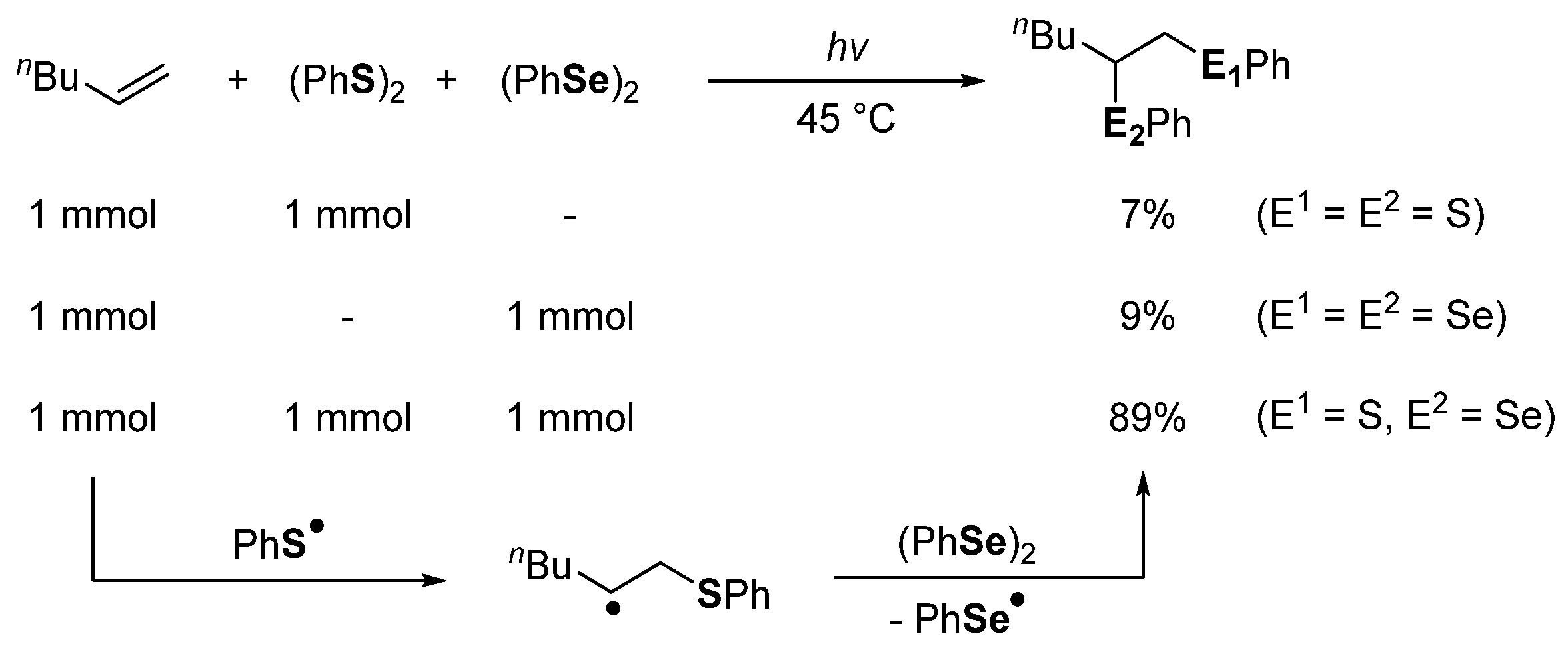{kind=link}
{kind=link}
{kind=link}
{kind=link}
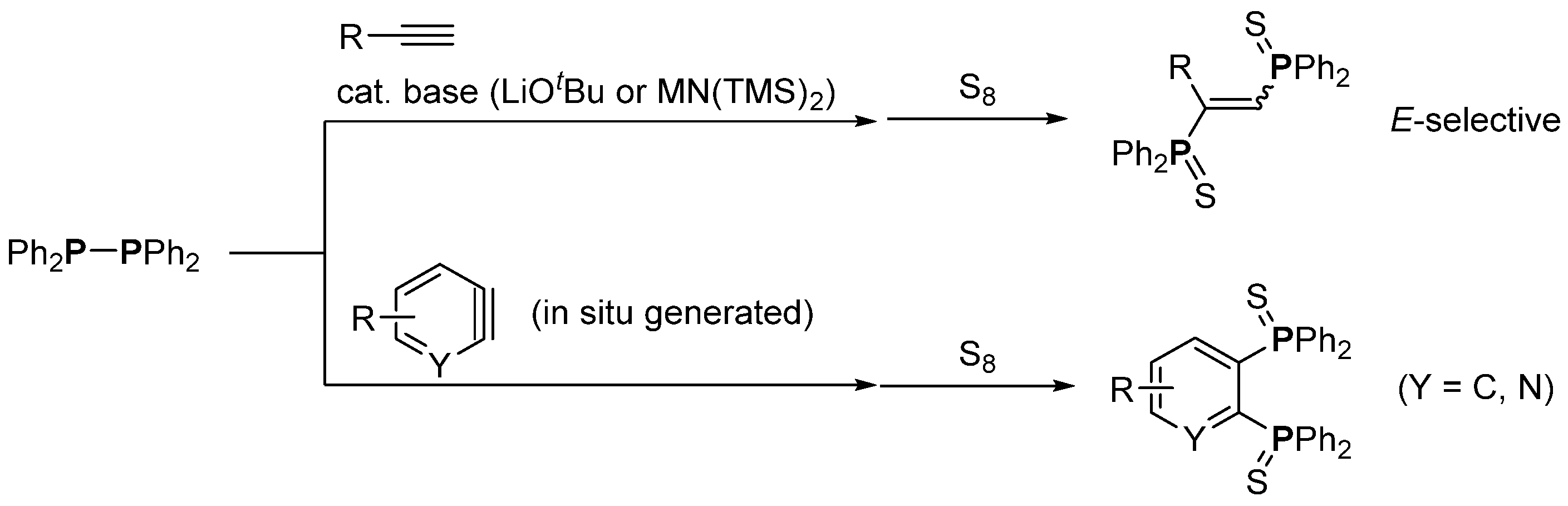{kind=link}
{kind=link}
{kind=link}
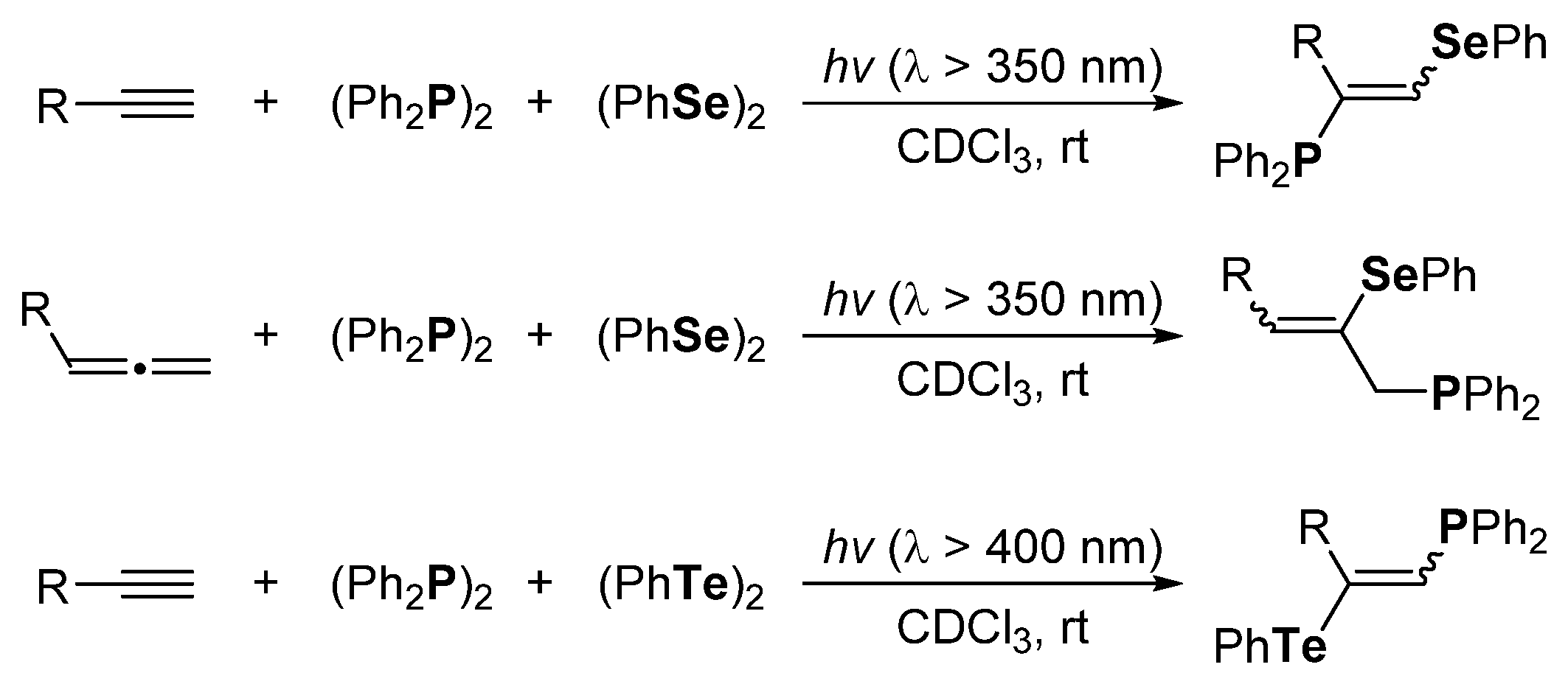{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
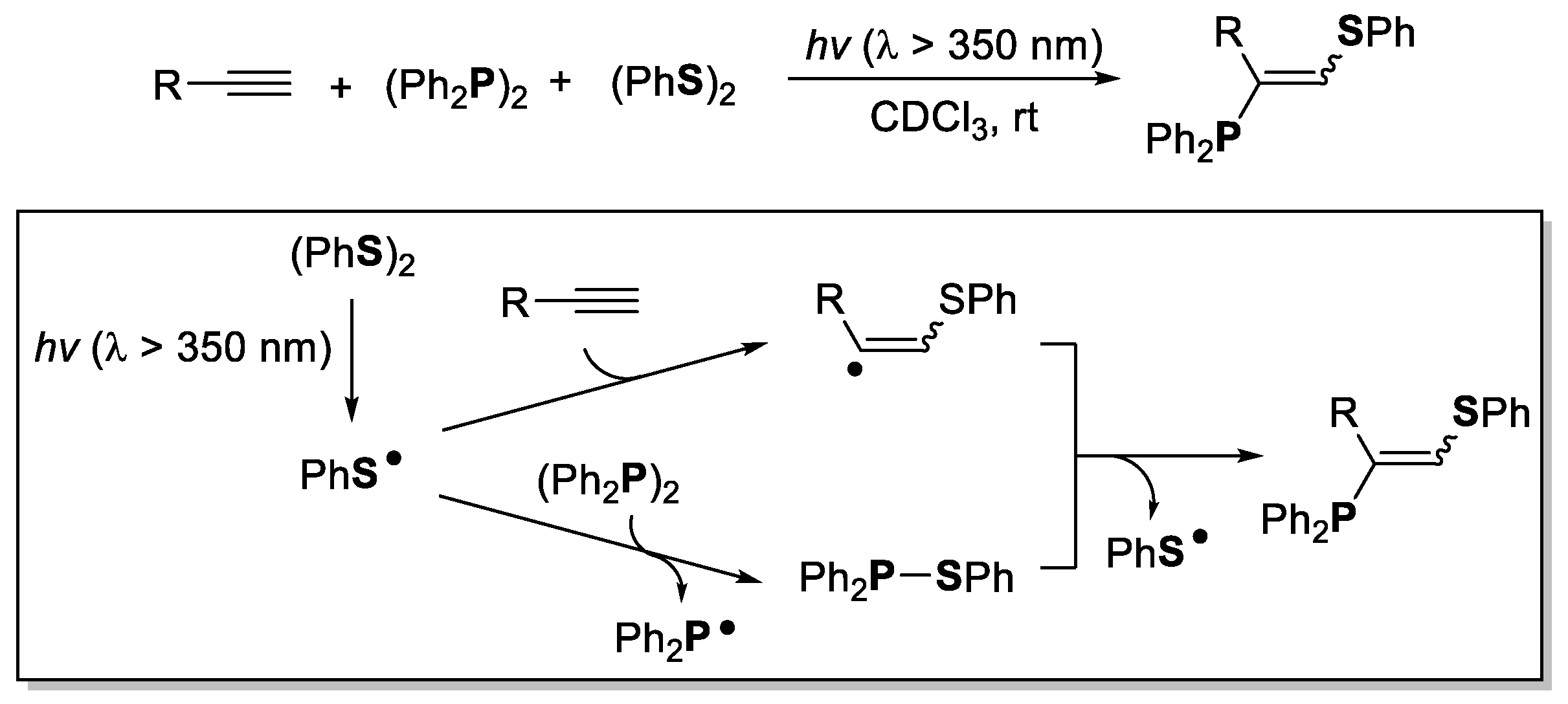
2. The Group 16 Element (O, S, Se, and Te)-Centered Interelement Compounds in Radical Reactions

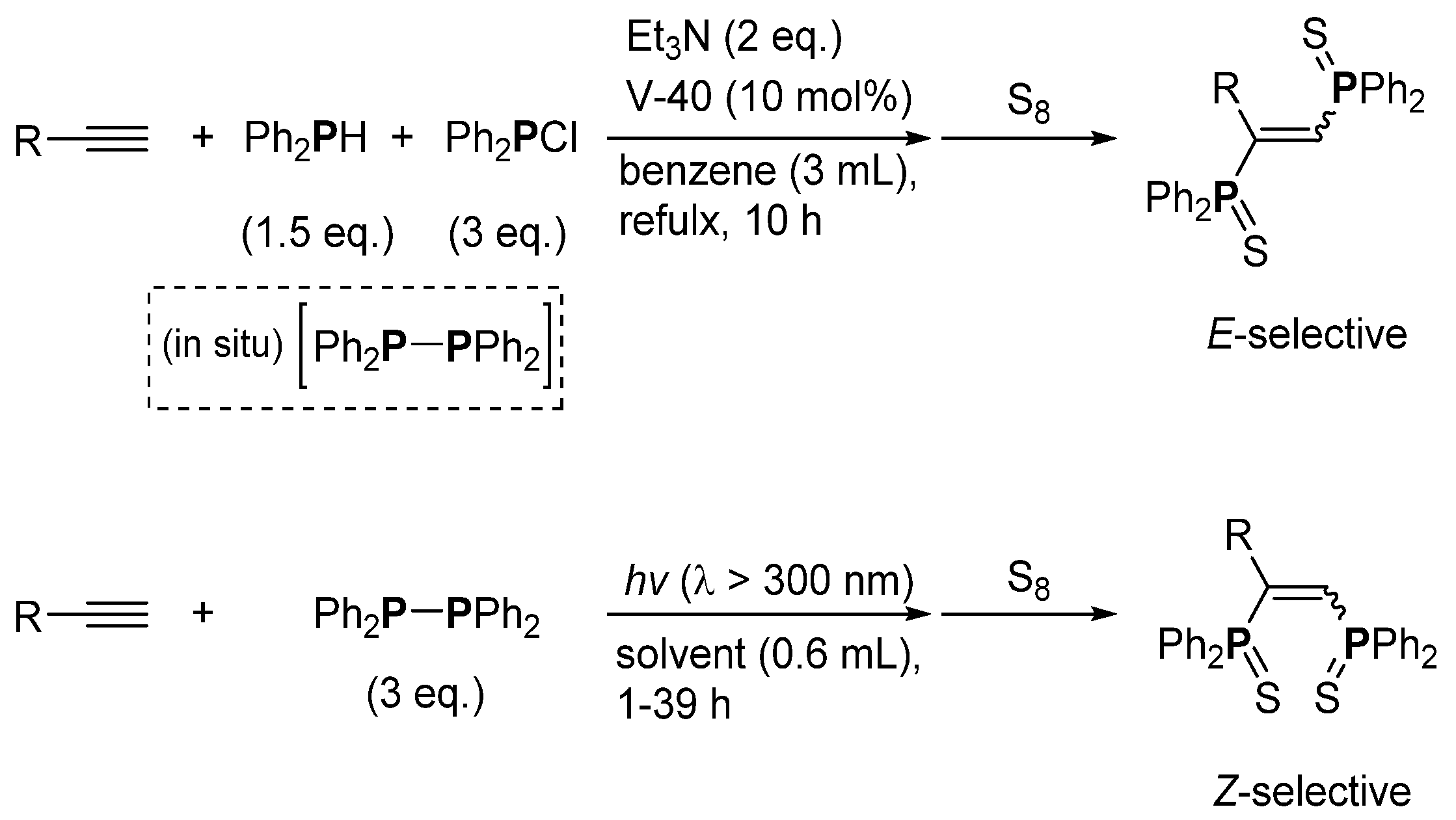
3. Phosphorus-Centered Interelement Compounds in Radical Reactions

4. Boron-Centered Interelement Compounds in Radical Addition Reactions


5. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Kakiuchi, F.; Kochi, T. Transition-Metal-Catalyzed Carbon-Carbon Bond Formation via Carbon-Hydrogen Bond Cleavage. Synthesis 2008, 2008, 3013–3039. [Google Scholar] [CrossRef]
- Achar, T.K.; Bose, A.; Mal, P. Mechanochemical synthesis of small organic molecules. Beilstein J. Org. Chem. 2017, 13, 1907–1931. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.; Liu, X.; Feng, X. Recent Advances in Metal-Catalyzed Asymmetric 1,4-Conjugate Addition (ACA) of Nonorganometallic Nucleophiles. Chem. Rev. 2018, 118, 7586–7656. [Google Scholar] [CrossRef]
- Hooshmand, S.E.; Heidari, B.; Sedghi, R.; Varma, R.S. Recent advances in the Suzuki–Miyaura cross-coupling reaction using efficient catalysts in eco-friendly media. Green Chem. 2019, 21, 381–405. [Google Scholar] [CrossRef]
- Samanta, R.; Matcha, K.; Antonchick, A.P. Metal-Free Oxidative Carbon-Heteroatom Bond Formation Through C-H Bond Functionalization. Eur. J. Org. Chem. 2013, 2013, 5769–5804. [Google Scholar] [CrossRef]
- Besset, T.; Poisson, T.; Pannecoucke, X. Direct Vicinal Difunctionalization of Alkynes: An Efficient Approach Towards the Synthesis of Highly Functionalized Fluorinated Alkenes. Eur. J. Org. Chem. 2015, 2015, 2765–2789. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.; Dong, Z. A Review on the Latest Progress of Chan-Lam Coupling Reaction. Adv. Synth. Catal. 2020, 362, 3311–3331. [Google Scholar] [CrossRef]
- Usman, M.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Recent Developments in Cobalt Catalyzed Carbon–Carbon and Carbon–Heteroatom Bond Formation via C–H Bond Functionalization. Synthesis 2017, 49, 1419–1443. [Google Scholar]
- Beletskaya, I.; Moberg, C. Element−Element Additions to Unsaturated Carbon−Carbon Bonds Catalyzed by Transition Metal Complexes. Chem. Rev. 2006, 106, 2320–2354. [Google Scholar] [CrossRef]
- Kawaguchi, S.-I.; Yamamoto, Y.; Ogawa, A. Catalytic synthesis of sulfur and phosphorus compounds via atom-economic reactions. Mendeleev Commun. 2020, 30, 129–138. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Ananikov, V.P. Transition-Metal-Catalyzed C–S, C–Se, and C–Te Bond Formations via Cross-Coupling and Atom-Economic Addition Reactions. Achievements and Challenges. Chem. Rev. 2022, 122, 16110–16293. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A. Addition of X−Y Reagents to Alkenes, Alkynes, and Allenes. In Comprehensive Organic Synthesis II; Elsevier: Amsterdam, The Netherlands, 2014; Volume 4, pp. 392–411. [Google Scholar]
- Liu, L.; Ward, R.M.; Schomaker, J.M. Mechanistic Aspects and Synthetic Applications of Radical Additions to Allenes. Chem. Rev. 2019, 119, 12422–12490. [Google Scholar] [CrossRef] [PubMed]
- Tamao, K.; Yamaguchi, S. Introduction to The Chemistry of Interelement Linkage. J. Organomet. Chem. 2000, 611, 3–4. [Google Scholar] [CrossRef]
- Tokitoh, N. New aspects in the chemistry of low-coordinated inter-element compounds of heavier Group 15 elements. J. Organomet. Chem. 2000, 611, 217–227. [Google Scholar] [CrossRef]
- Hayashi, T.; Matsumoto, Y.; Ito, Y. Palladium-Catalyzed Asymmetric 1,4-Disilylation of α,β-Unsaturated Ketones: Catalytic Asymmetric Synthesis of β-Hydroxy Ketones. J. Am. Chem. Soc. 1988, 110, 5579–5581. [Google Scholar] [CrossRef]
- Lu, A.; Ji, X.; Zhou, B.; Wu, Z.; Zhang, Y. Palladium-Catalyzed, C−H Silylation through Palladacycles Generated from Aryl Halides. Angew. Chem. Int. Ed. 2018, 57, 3233–3237. [Google Scholar] [CrossRef]
- Wollenburg, M.; Bajohr, J.; Marchese, A.D.; Whyte, A.; Glorius, F.; Lautens, M. Palladium-Catalyzed Disilylation and Digermanylation of Alkene Tethered Aryl Halides: Direct Access to Versatile Silylated and Germanylated Heterocycles. Org. Lett. 2020, 22, 3679–3683. [Google Scholar] [CrossRef]
- Louka, A.; Stratakis, M. Synthesis of Vinylgermanes via the Au/TiO2-Catalyzed cis-1,2-Digermylation of Alkynes and the Regioselective Hydrogermylation of Allenes. Org. Lett. 2021, 23, 3599–3603. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Yokoyama, H.; Sekiguchi, M.; Kambe, N.; Sonoda, N. Photo-initiated addition of diphenyl diselenide to allenes. Tetrahedron Lett. 1990, 31, 5931–5934. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, H.; Yokoyama, K.; Masawaki, T.; Kambe, N.; Sonoda, N. Photo-initiated addition of diphenyl diselenide to acetylenes. J. Org. Chem. 1991, 56, 5721–5723. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Yokoyama, H.; Obayashi, R.; Kambe, N.; Sonoda, N. Photo-initiated addition of diphenyl ditelluride to acetylenes. J. Chem. Soc. Chem. Commun. 1991, 1748–1750. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Obayashi, R.; Han, L.-B.; Kambe, N.; Sonoda, N. Photo-Induced Ditelluration of Acetylenes with Diphenyl Ditelluride. Tetrahedron 1993, 49, 1177–1188. [Google Scholar] [CrossRef]
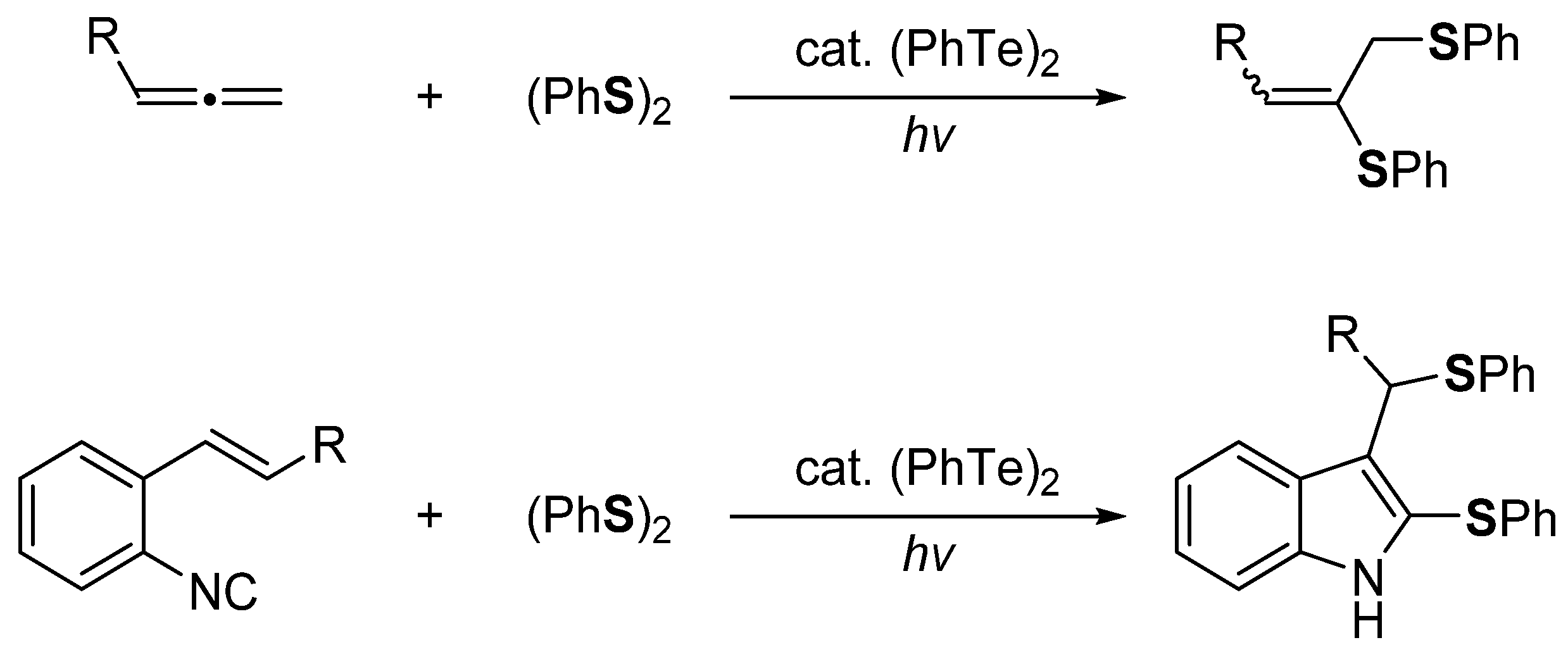
- Tran, C.C.; Kawaguchi, S.-I.; Sato, F.; Nomoto, A.; Ogawa, A. Photoinduced Cyclizations of o-Diisocyanoarenes with Organic Diselenides and Thiols that Afford Chalcogenated Quinoxalines. J. Org. Chem. 2020, 85, 7258–7266. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Iwata, K.; Nomoto, A.; Ogawa, A. Photochemical intramolecular cyclization of o-alkynylaryl isocyanides with organic dichalcogenides leading to 2,4-bischalcogenated quinolines. Org. Biomol. Chem. 2011, 9, 3768–3775. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Iwata, K.; Ogawa, A. (PhTe)2-Mediated Intramolecular Radical Cyclization of o-Ethynylaryl Isocyanides Leading to Bistellurated Quinolines upon Visible-Light Irradiation. Org. Lett. 2009, 11, 3422–3424. [Google Scholar] [CrossRef]
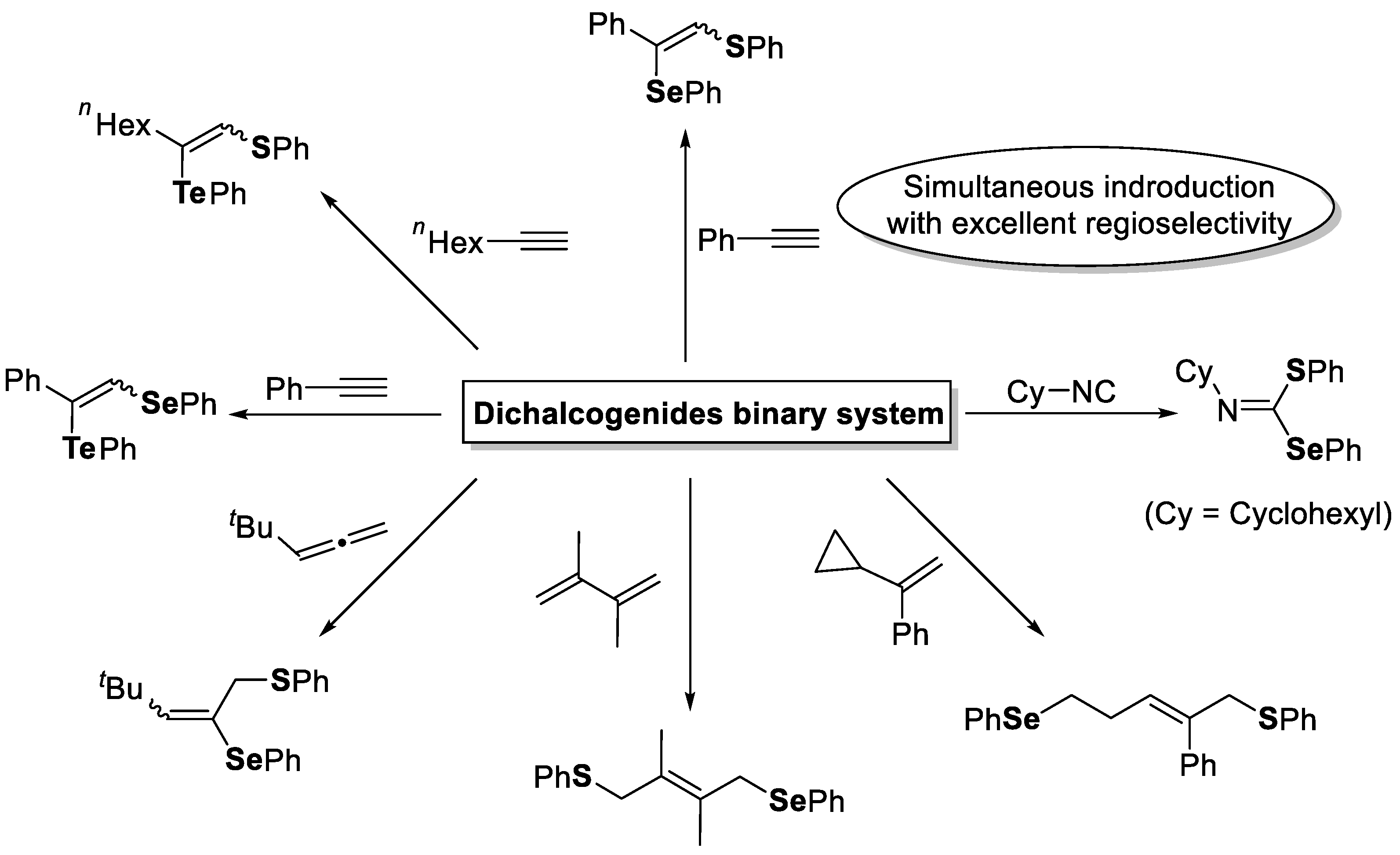
- Ogawa, A.; Tanaka, H.; Yokoyama, H.; Obayashi, R.; Yokoyama, K.; Sonoda, N. A highly selective thioselenation of olefins using disulfide-diselenide mixed system. J. Org. Chem. 1992, 57, 111–115. [Google Scholar] [CrossRef]
- Ogawa, A.; Obayashi, R.; Ine, H.; Tsuboi, Y.; Sonoda, N.; Hirao, T. Highly Regioselective Thioselenation of Acetylenes by Using a (PhS)2−(PhSe)2 Binary System. J. Org. Chem. 1998, 63, 881–884. [Google Scholar] [CrossRef]
- Ogawa, A.; Obayashi, R.; Doi, M.; Sonoda, N.; Hirao, T. A Novel Photoinduced Thioselenation of Allenes by Use of a Disulfide−Diselenide Binary System. J. Org. Chem. 1998, 63, 4277–4281. [Google Scholar] [CrossRef]
- Ogawa, A.; Obayashi, R.; Sonoda, N.; Hirao, T. Diphenyl diselenide-assisted dithiolation of 1,3-dienes with diphenyl disulfide upon irradiation with near-UV light. Tetrahedron Lett. 1998, 39, 1577–1578. [Google Scholar] [CrossRef]
- Ogawa, A.; Ogawa, I.; Obayashi, R.; Umezu, K.; Doi, M.; Hirao, T. Highly Selective Thioselenation of Vinylcyclopropanes with a (PhS)2−(PhSe)2 Binary System and Its Application to Thiotelluration. J. Org. Chem. 1999, 64, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Tsuchii, K.; Tsuboi, Y.; Kawaguchi, S.-I.; Takahashi, J.; Sonoda, N.; Nomoto, A.; Ogawa, A. Highly Selective Double Chalcogenation of Isocyanides with Disulfide−Diselenide Mixed Systems. J. Org. Chem. 2006, 72, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Mitamura, T.; Iwata, K.; Ogawa, A. Photoinduced Intramolecular Cyclization of o-Ethenylaryl Isocyanides with Organic Disulfides Mediated by Diphenyl Ditelluride. J. Org. Chem. 2011, 76, 3880–3887. [Google Scholar] [CrossRef]
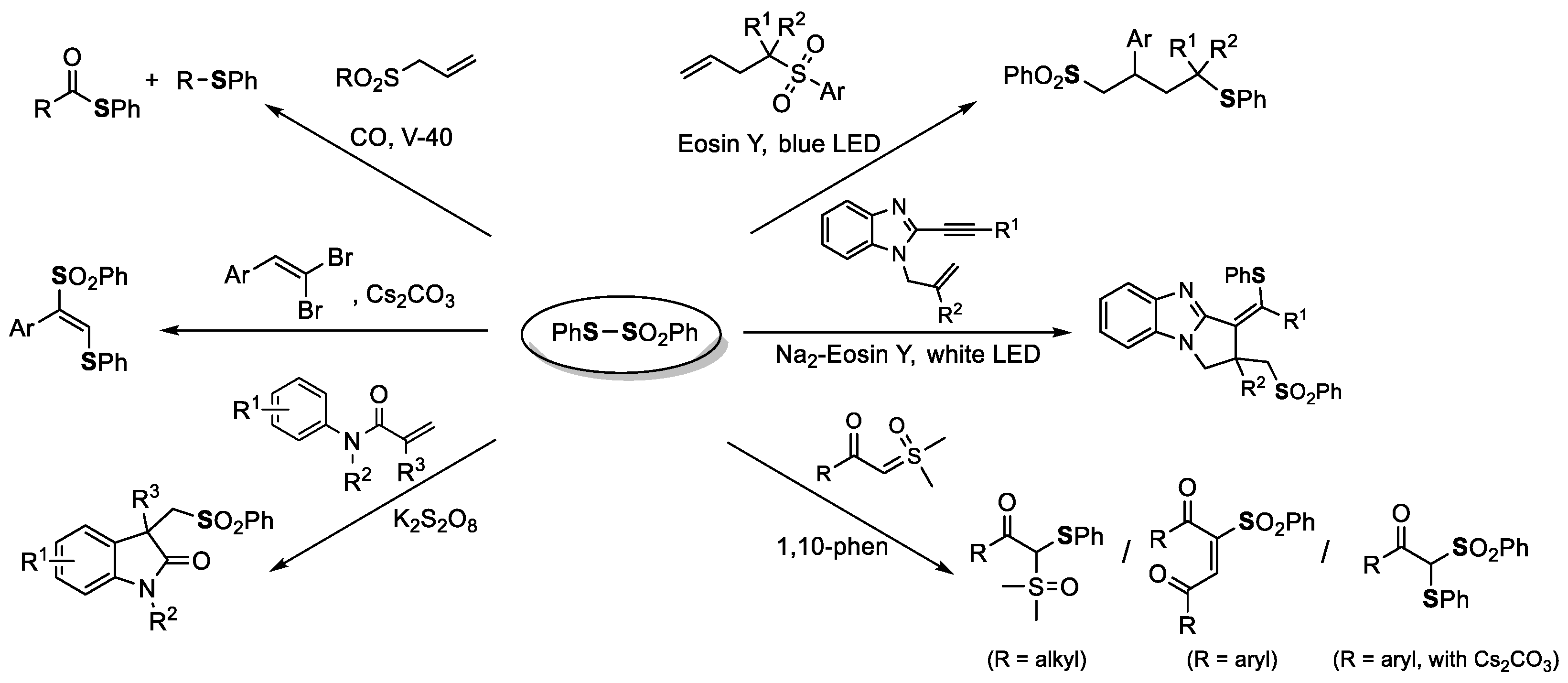
- Kim, S.; Kim, S.; Otsuka, N.; Ryu, I. Tin-Free Radical Carbonylation: Thiol Ester Synthesis Using Alkyl Allyl Sulfone Precursors, Phenyl Benzenethiosulfonate, and CO. Angew. Chem. Int. Ed. 2005, 44, 6183–6186. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Z.; Ji, P.-Y.; Liu, Y.-F.; Xu, J.-W.; Guo, C.-C. Disulfides as Sulfonylating Precursors for the Synthesis of Sulfone-Contain ing Oxindoles. Adv. Synth. Catal. 2016, 358, 2976–2983. [Google Scholar] [CrossRef]
- Reddy, R.J.; Kumari, A.H.; Kumar, J.J.; Nanubolu, J.B. Cs2CO3-Mediated Vicinal Thiosulfonylation of 1,1-Dibromo-1-Alkenes with Thiosulfonates: An Expedient Synthesis of (E)-1,2-Thiosulfonylethenes. Adv. Synth. Catal. 2019, 361, 1587–1591. [Google Scholar] [CrossRef]
- Wang, F.; Liu, B.-X.; Rao, W.; Wang, S.-Y. Metal-Free Chemoselective Reaction of Sulfoxonium Ylides and Thiosulfonates: Diverse Synthesis of 1,4-Diketones, Aryl Sulfursulfoxonium Ylides, and β-Keto Thiosulfones Derivatives. Org. Lett. 2020, 22, 6600–6604. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, N.; Xu, Y.; Chen, Y. Visible-Light-Induced Radical Cascade Reaction of 1-Allyl-2-ethynylbenzoimidazoles with Thiosulfonates to Assemble Thiosulfonylated Pyrrolo[1,2-a]benzimidazoles. J. Org. Chem. 2021, 86, 16882–16891. [Google Scholar] [CrossRef]
- Liu, X.-Y.; Tian, S.-Y.; Jiang, Y.-F.; Rao, W.; Wang, S.-Y. Visible-Light-Triggered Sulfonylation/Aryl Migration/Desulfonylation and C–S/Se Bond Formation Reaction: 1,2,4-Trifunctionalization of Butenyl Benzothiazole Sulfone with Thiosulfonate/Selenosulfonates. Org. Lett. 2021, 23, 8246–8251. [Google Scholar] [CrossRef]
- Tamai, T.; Nomoto, A.; Tsuchii, K.; Minamida, Y.; Mitamura, T.; Sonoda, M.; Ogawa, A. Highly selective perfluoroalkylchalcogenation of alkynes by the combination of iodoperfluoroalkanes and organic dichalcogenides upon photoirradiation. Tetrahedron 2012, 68, 10516–10522. [Google Scholar] [CrossRef]
- Kuchen, W.; Buchwald, H. Zur Kenntnis der Organophosphorverbindungen, II. Das Tetraphenyldiphosphin. Chem. Ber. 1958, 91, 2871–2877. [Google Scholar] [CrossRef]
- Blake, A.; McQuillan, G.; Oxton, I.; Troy, D. Diphosphine derivatives: Part VIII. Tetraphenyldiphosphine dioxide and tetraphenyldiphosphine disulphide: Structure and skeletal vibrations. Preparation of diphenylphos-ph. J. Mol. Struct. 1982, 78, 265–271. [Google Scholar] [CrossRef]
- Hirano, K.; Miura, M. Recent advances in diphosphination of alkynes and alkenes. Tetrahedron Lett. 2017, 58, 4317–4322. [Google Scholar] [CrossRef]
- Dodds, D.L.; Haddow, M.F.; Orpen, A.G.; Pringle, P.G.; Woodward, G. Stereospecific Diphosphination of Activated Acetylenes: A General Route to Backbone-Functionalized, Chelating 1,2-Diphosphinoethenes. Organometallics 2006, 25, 5937–5945. [Google Scholar] [CrossRef]
- Okugawa, Y.; Hirano, K.; Miura, M. Brønsted Base Mediated Stereoselective Diphosphination of Terminal Alkynes with Diphosphanes. Org. Lett. 2017, 19, 2973–2976. [Google Scholar] [CrossRef] [PubMed]
- Okugawa, Y.; Hayashi, Y.; Kawauchi, S.; Hirano, K.; Miura, M. Diphosphination of Arynes with Diphosphines. Org. Lett. 2018, 20, 3670–3673. [Google Scholar] [CrossRef]
- Yorimitsu, H. Homolytic substitution at phosphorus for C–P bond formation in organic synthesis. Beilstein J. Org. Chem. 2013, 9, 1269–1277. [Google Scholar] [CrossRef]
- Kawaguchi, S.-I.; Ogawa, A. Applications of Diphosphines in Radical Reactions. Asian J. Org. Chem. 2019, 8, 1164–1173. [Google Scholar] [CrossRef]
- Hewertson, W.; Taylor, I.C. The reaction of tetramethyldiphosphine with butadiene. J. Chem. Soc. C. 1970, 1990–1992. [Google Scholar] [CrossRef]
- Tzschach, A.; Baensch, S. Organoarsen-Verbindungen. XVII. Zur Umsetzung von Biphosphinen und Biarsinen mit Phenylacetylen. J. Prakt. Chem. 1971, 313, 254–258. [Google Scholar] [CrossRef]
- Otomura, N.; Okugawa, Y.; Hirano, K.; Miura, M. vic-Diphosphination of Alkenes with Silylphosphine under Visible-Light-Promoted Photoredox Catalysis. Org. Lett. 2017, 19, 4802–4805. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Yorimitsu, H.; Oshima, K. Synthesis of (E)-1,2-Diphosphanylethene Derivatives from Alkynes by Radical Addition of Tetraorganodiphosphane Generated In Situ. Angew. Chem. Int. Ed. 2005, 44, 1694–1696. [Google Scholar] [CrossRef] [PubMed]
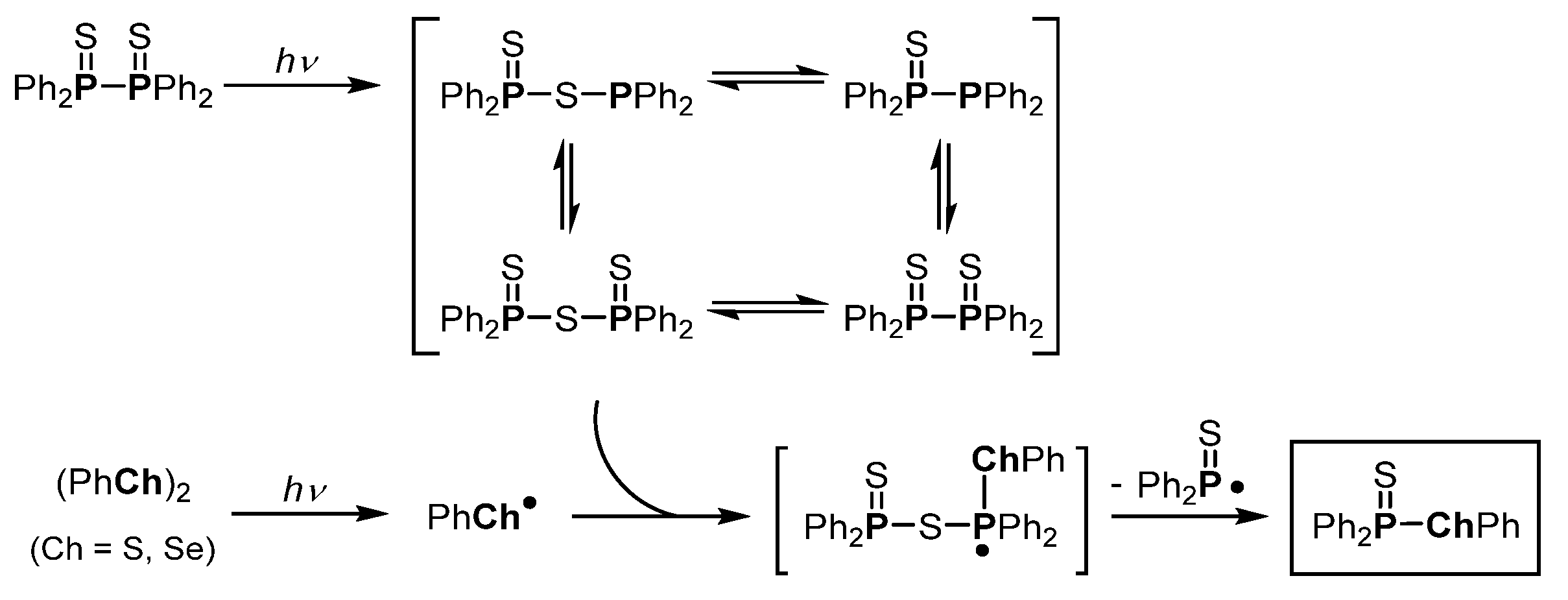
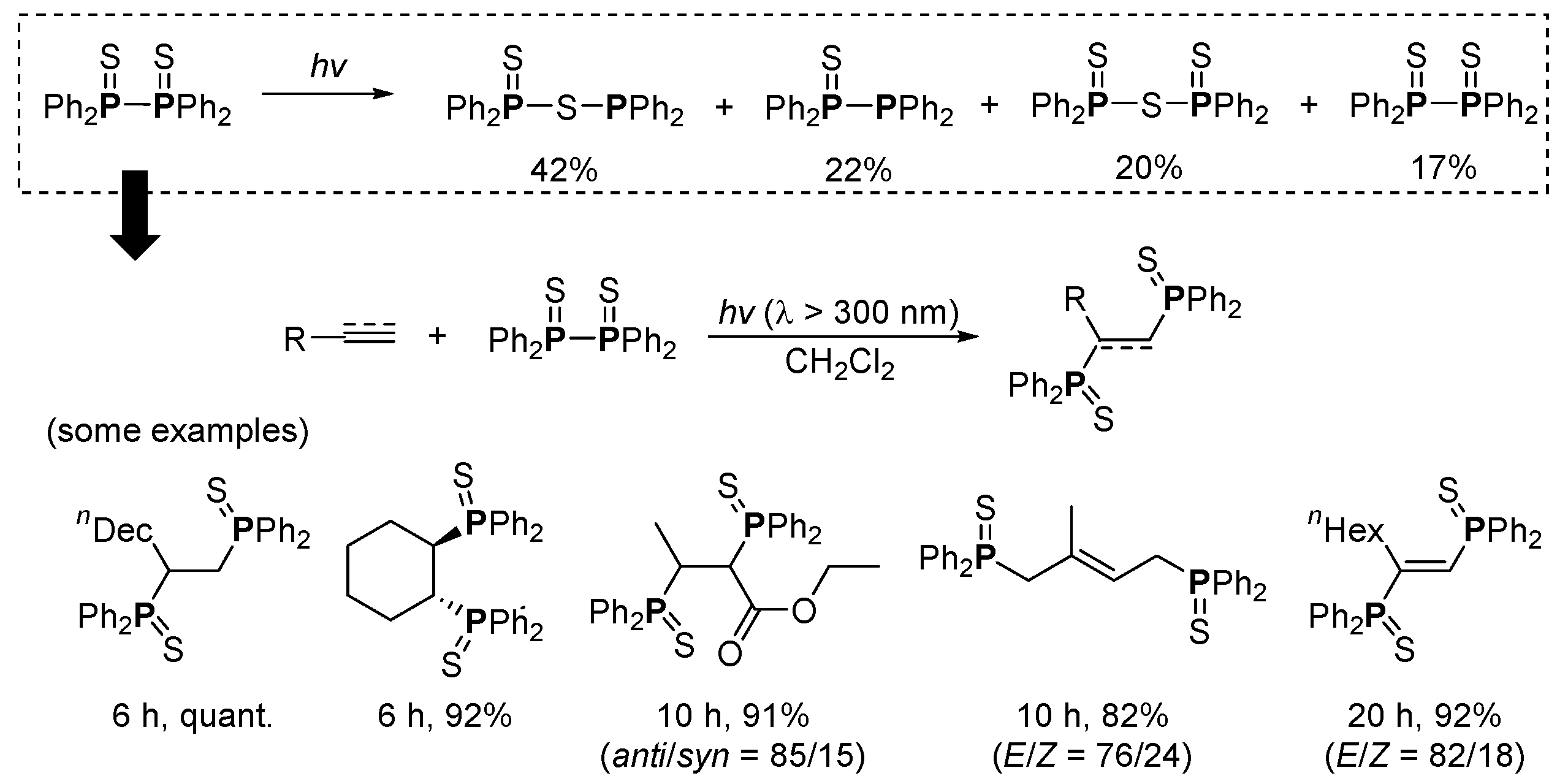
- Kawaguchi, S.-I.; Nagata, S.; Shirai, T.; Tsuchii, K.; Nomoto, A.; Ogawa, A. Photochemical behaviors of tetraphenyldiphosphine in the presence of alkynes. Tetrahedron Lett. 2006, 47, 3919–3922. [Google Scholar] [CrossRef]
- Shirai, T.; Kawaguchi, S.-i.; Nomoto, A.; Ogawa, A. Photoinduced highly selective thiophosphination of alkynes using a (PhS)2/(Ph2P)2 binary system. Tetrahedron Lett. 2008, 49, 4043–4046. [Google Scholar] [CrossRef]
- Wada, T.; Kondoh, A.; Yorimitsu, H.; Oshima, K. Intermolecular Radical Addition of Alkylthio- and Arylthiodiphenylphosphines to Terminal Alkynes. Org. Lett. 2008, 10, 1155–1157. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Yorimitsu, H.; Oshima, K. Regio- and stereoselective synthesis of 1-aryl-1-thio-2-thiophosphinylethene derivatives via a radical process. Tetrahedron 2009, 65, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Kawaguchi, S.-I.; Shirai, T.; Ohe, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. Highly Regioselective Simultaneous Introduction of Phosphino and Seleno Groups into Unsaturated Bonds by the Novel Combination of (Ph2P)2 and (PhSe)2 upon Photoirradiation. J. Org. Chem. 2009, 74, 1751–1754. [Google Scholar] [CrossRef]
- Kawaguchi, S.-i.; Ohe, T.; Shirai, T.; Nomoto, A.; Sonoda, M.; Ogawa, A. Highly Selective Phosphinotelluration of Terminal Alkynes Using a (Ph2P)2−(PhTe)2 Mixed System upon Visible Light Irradiation: Straightforward Access to 1-Phosphino-2-telluro-alkenes. Organometallics 2010, 29, 312–316. [Google Scholar] [CrossRef]
- Sato, Y.; Kawaguchi, S.-I.; Nomoto, A.; Ogawa, A. Highly Selective Phosphinylphosphination of Alkenes with Tetraphenyldiphosphine Monoxide. Angew. Chem. Int. Ed. 2016, 55, 9700–9703. [Google Scholar] [CrossRef]
- Takano, H.; Katsuyama, H.; Hayashi, H.; Kanna, W.; Harabuchi, Y.; Maeda, S.; Mita, T. A theory-driven synthesis of symmetric and unsymmetric 1,2-bis(diphenylphosphino)ethane analogues via radical difunctionalization of ethylene. Nat. Commun. 2022, 13, 7034. [Google Scholar] [CrossRef]
- Tran, D.P.; Sato, Y.; Yamamoto, Y.; Kawaguchi, S.-I.; Kodama, S.; Nomoto, A.; Ogawa, A. Highly regio- and stereoselective phosphinylphosphination of terminal alkynes with tetraphenyldiphosphine monoxide under radical conditions. Beilstein J. Org. Chem. 2021, 17, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Kawaguchi, S.-I.; Nomoto, A.; Ogawa, A. Synthesis of Bis(phosphanyl)alkane Monosulfides by the Addition of Diphosphane Monosulfides to Alkenes under Light. Chem. A Eur. J. 2018, 25, 2295–2302. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Tanaka, R.; Kodama, S.; Nomoto, A.; Ogawa, A. Photoinduced Bisphosphination of Alkynes with Phosphorus Interelement Compounds and Its Application to Double-Bond Isomerization. Molecules 2022, 27, 1284. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, S.-I.; Ogawa, A.; Sato, Y.; Nomoto, A. Photoinduced Coupling Reaction of Diphenyl(2,4,6-trimethylbenzoyl)phosphine Oxide with Interelement Compounds: Application to the Synthesis of Thio- or Selenophosphinates. Synthesis 2017, 49, 3558–3567. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, Y.; Tanaka, R.; Ota, M.; Nishimura, M.; Tran, C.C.; Kawaguchi, S.-I.; Kodama, S.; Nomoto, A.; Ogawa, A. Photoinduced Syntheses and Reactivities of Phosphorus-Containing Interelement Compounds. J. Org. Chem. 2020, 85, 14708–14719. [Google Scholar] [CrossRef]
- Sato, Y.; Nishimura, M.; Kawaguchi, S.; Nomoto, A.; Ogawa, A. Reductive Rearrangement of Tetraphenyldiphosphine Disulfide To Trigger the Bisthiophosphinylation of Alkenes and Alkynes. Chem. A Eur. J. 2019, 25, 6797–6806. [Google Scholar] [CrossRef]
- Cuenca, A.B.; Shishido, R.; Ito, H.; Fernández, E. Transition-metal-free B–B and B–interelement reactions with organic molecules. Chem. Soc. Rev. 2017, 46, 415–430. [Google Scholar] [CrossRef]
- Yan, G.; Huang, D.; Wu, X. Recent Advances in C-B Bond Formation through a Free Radical Pathway. Adv. Synth. Catal. 2017, 360, 1040–1053. [Google Scholar] [CrossRef]
- Nguyen, V.D.; Nguyen, V.; Jin, S.; Dang, H.T.; Larionov, O.V. Organoboron chemistry comes to light: Recent advances in photoinduced synthetic approaches to organoboron compounds. Tetrahedron 2018, 75, 584–602. [Google Scholar] [CrossRef]
- Friese, F.W.; Studer, A. New avenues for C–B bond formation via radical intermediates. Chem. Sci. 2019, 10, 8503–8518. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.-M.; Guo, X.-N.; Braunschweig, H.; Radius, U.; Marder, T.B. Photoinduced Borylation for the Synthesis of Organoboron Compounds. Chem. Rev. 2021, 121, 3561–3597. [Google Scholar] [CrossRef] [PubMed]
- Yadagiri, B.; Daipule, K.; Singh, S.P. Photoinduced Borylation Reactions: An Overview. Asian, J. Org. Chem. 2020, 10, 7–37. [Google Scholar] [CrossRef]
- Lai, D.; Ghosh, S.; Hajra, A. Light-induced borylation: Developments and mechanistic insights. Org. Biomol. Chem. 2021, 19, 4397–4428. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, R.D.; Neeve, E.C.; Braunschweig, H.; Marder, T.B. sp2–sp3 diboranes: Astounding structural variability and mild sources of nucleophilic boron for organic synthesis. Chem. Commun. 2015, 51, 9594–9607. [Google Scholar] [CrossRef]
- Neeve, E.C.; Geier, S.J.; Mkhalid, I.A.I.; Westcott, S.A.; Marder, T.B. Diboron(4) Compounds: From Structural Curiosity to Synthetic Workhorse. Chem. Rev. 2016, 116, 9091–9161. [Google Scholar] [CrossRef] [Green Version]
- Ceron, P.; Finch, A.; Frey, J.; Kerrigan, J.; Parsons, T.; Urry, G.; Schlesinger, H. Diboron Tetrachloride and Tetrafluoride as Reagents for the Synthesis of Organoboron Compounds. II. The Behavior of the Diboron Tetrahalides toward Unsaturated Organic Compounds1. J. Am. Chem. Soc. 1959, 81, 6368–6371. [Google Scholar] [CrossRef]
- Rudolph, R.W. Mechanism of addition of diboron tetrachloride to unsaturated organic compounds. J. Am. Chem. Soc. 1967, 89, 4216–4217. [Google Scholar] [CrossRef]
- Zeldin, M.; Gatti, A.R.; Wartik, T. Stereochemical investigations of the addition of diboron tetrachloride to unsaturated organic molecules. J. Am. Chem. Soc. 1967, 89, 4217–4218. [Google Scholar] [CrossRef]
- Bonet, A.; Pubill-Ulldemolins, C.; Bo, C.; Gulyás, H.; Fernández, E. Transition-Metal-Free Diboration Reaction by Activation of Diboron Compounds with Simple Lewis Bases. Angew. Chem. Int. Ed. 2011, 50, 7158–7161. [Google Scholar] [CrossRef]
- Bonet, A.; Gulyás, H.; Fernández, E. Metal-Free Catalytic Boration at the β-Position of α,β-Unsaturated Compounds: A Challenging Asymmetric Induction. Angew. Chem. Int. Ed. 2010, 49, 5130–5134. [Google Scholar] [CrossRef]
- Pubill-Ulldemolins, C.; Bonet, A.; Bo, C.; Gulyás, H.; Fernández, E. Activation of Diboron Reagents with Brønsted Bases and Alcohols: An Experimental and Theoretical Perspective of the Organocatalytic Boron Conjugate Addition Reaction. Chem. A Eur. J. 2012, 18, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Pubill-Ulldemolins, C.; Bonet, A.; Gulyás, H.; Bo, C.; Fernández, E. Essential role of phosphines in organocatalytic β-boration reaction. Org. Biomol. Chem. 2012, 10, 9677–9682. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, Y.; Hirano, K.; Takita, R.; Uchiyama, M. Trans-Diborylation of Alkynes: Pseudo-Intramolecular Strategy Utilizing a Propargylic Alcohol Unit. J. Am. Chem. Soc. 2014, 136, 8532–8535. [Google Scholar] [CrossRef] [PubMed]
- Nagao, K.; Ohmiya, H.; Sawamura, M. Anti-Selective Vicinal Silaboration and Diboration of Alkynoates through Phosphine Organocatalysis. Org. Lett. 2015, 17, 1304–1307. [Google Scholar] [CrossRef]
- Morinaga, A.; Nagao, K.; Ohmiya, H.; Sawamura, M. Synthesis of 1,1-Diborylalkenes through a Brønsted Base Catalyzed Reaction between Terminal Alkynes and Bis(pinacolato)diboron. Angew. Chem. Int. Ed. 2015, 54, 15859–15862. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.; Liu, G.; Huang, Z. Mixed Diboration of Alkynes Catalyzed by LiOH: Regio- and Stereoselective Synthesis of cis-1,2-Diborylalkenes. Org. Lett. 2018, 20, 7363–7366. [Google Scholar] [CrossRef]
- Kuang, Z.; Gao, G.; Song, Q. Base-catalyzed diborylation of alkynes: Synthesis and applications of cis-1,2-bis(boryl)alkenes. Sci. China Chem. 2019, 62, 62–66. [Google Scholar] [CrossRef]
- Liu, X.; Ming, W.; Luo, X.; Friedrich, A.; Maier, J.; Radius, U.; Santos, W.L.; Marder, T.B. Regio- and Stereoselective Synthesis of 1,1-Diborylalkenes via Brønsted Base-Catalyzed Mixed Diboration of Alkynyl Esters and Amides with BpinBdan. Eur. J. Org. Chem. 2020, 2020, 1941–1946. [Google Scholar] [CrossRef]
- Farre, A.; Soares, K.; Briggs, R.A.; Balanta, A.; Benoit, D.M.; Bonet, A. Amine Catalysis for the Organocatalytic Diboration of Challenging Alkenes. Chem. A Eur. J. 2016, 22, 17552–17556. [Google Scholar] [CrossRef]
- Ohmura, T.; Morimasa, Y.; Suginome, M. 4,4′-Bipyridine-catalyzed Stereoselective trans-Diboration of Acetylenedicarboxylates to 2,3-Diborylfumarates. Chem. Lett. 2017, 46, 1793–1796. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Duan, Y.; Shen, Y.; Martin, R. Stereoselective Base-Catalyzed 1,1-Silaboration of Terminal Alkynes. Angew. Chem. Int. Ed. 2020, 59, 2061–2065. [Google Scholar] [CrossRef] [PubMed]
- Kojima, C.; Lee, K.-H.; Lin, Z.; Yamashita, M. Direct and Base-Catalyzed Diboration of Alkynes Using the Unsymmetrical Diborane(4), pinB-BMes2. J. Am. Chem. Soc. 2016, 138, 6662–6669. [Google Scholar] [CrossRef] [PubMed]
- Yukimori, D.; Nagashima, Y.; Wang, C.; Muranaka, A.; Uchiyama, M. Quadruple Borylation of Terminal Alkynes. J. Am. Chem. Soc. 2019, 141, 9819–9822. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Kojima, C.; Lee, K.-H.; Morisako, S.; Lin, Z.; Yamashita, M. Mechanistic study on the reaction of pinB-BMes2 with alkynes based on experimental investigation and DFT calculations: Gradual change of mechanism depending on the substituent. Chem. Sci. 2021, 12, 9806–9815. [Google Scholar] [CrossRef]
- Verma, A.; Snead, R.F.; Dai, Y.; Slebodnick, C.; Yang, Y.; Yu, H.; Yao, F.; Santos, W.L. Substrate-Assisted, Transition-Metal-Free Diboration of Alkynamides with Mixed Diboron: Regio- and Stereoselective Access to trans-1,2-Vinyldiboronates. Angew. Chem. Int. Ed. 2017, 56, 5111–5115. [Google Scholar] [CrossRef]
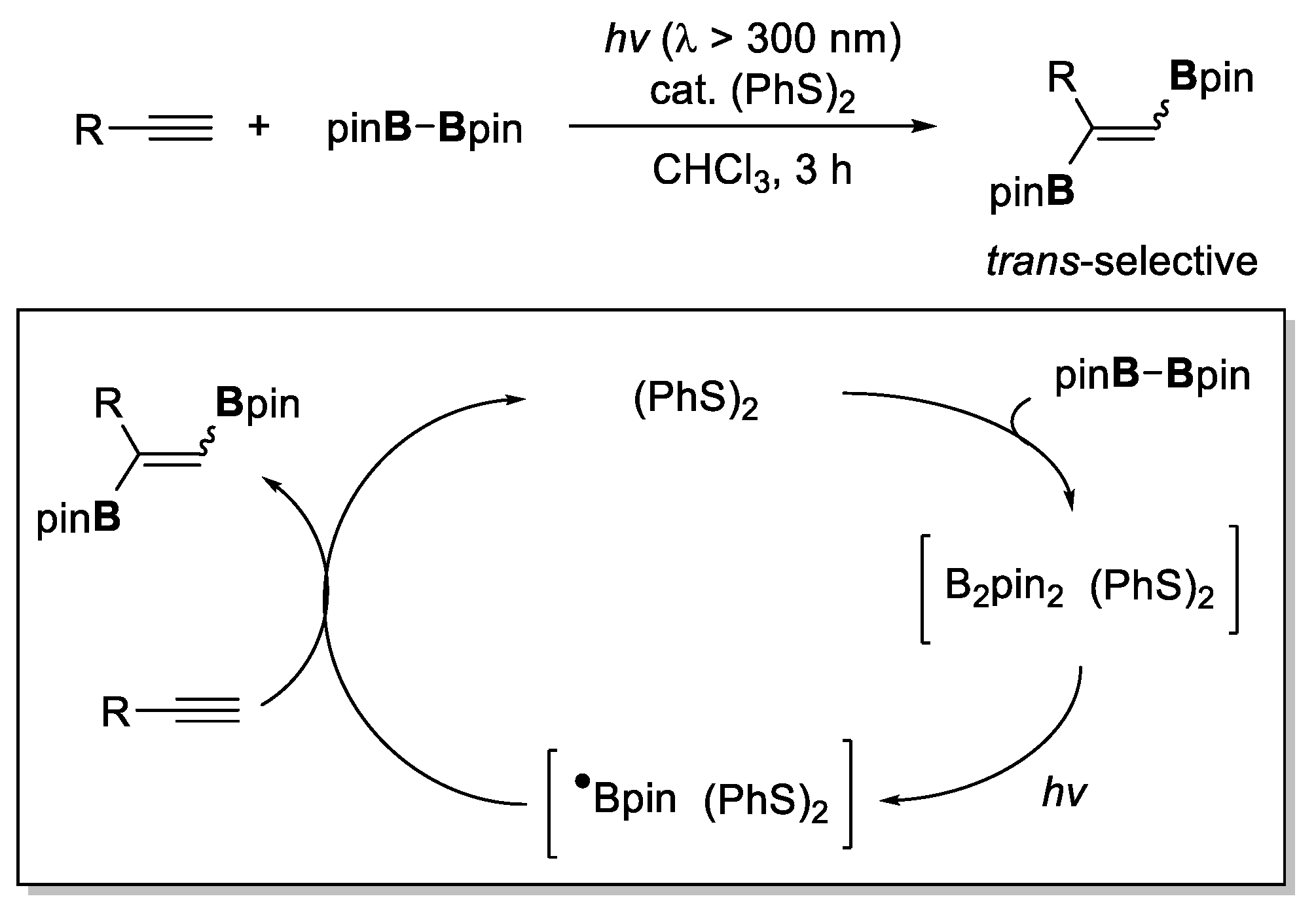
- Yoshimura, A.; Takamachi, Y.; Han, L.; Ogawa, A. Organosulfide-Catalyzed Diboration of Terminal Alkynes under Light. Chem. A Eur. J. 2015, 21, 13930–13933. [Google Scholar] [CrossRef]
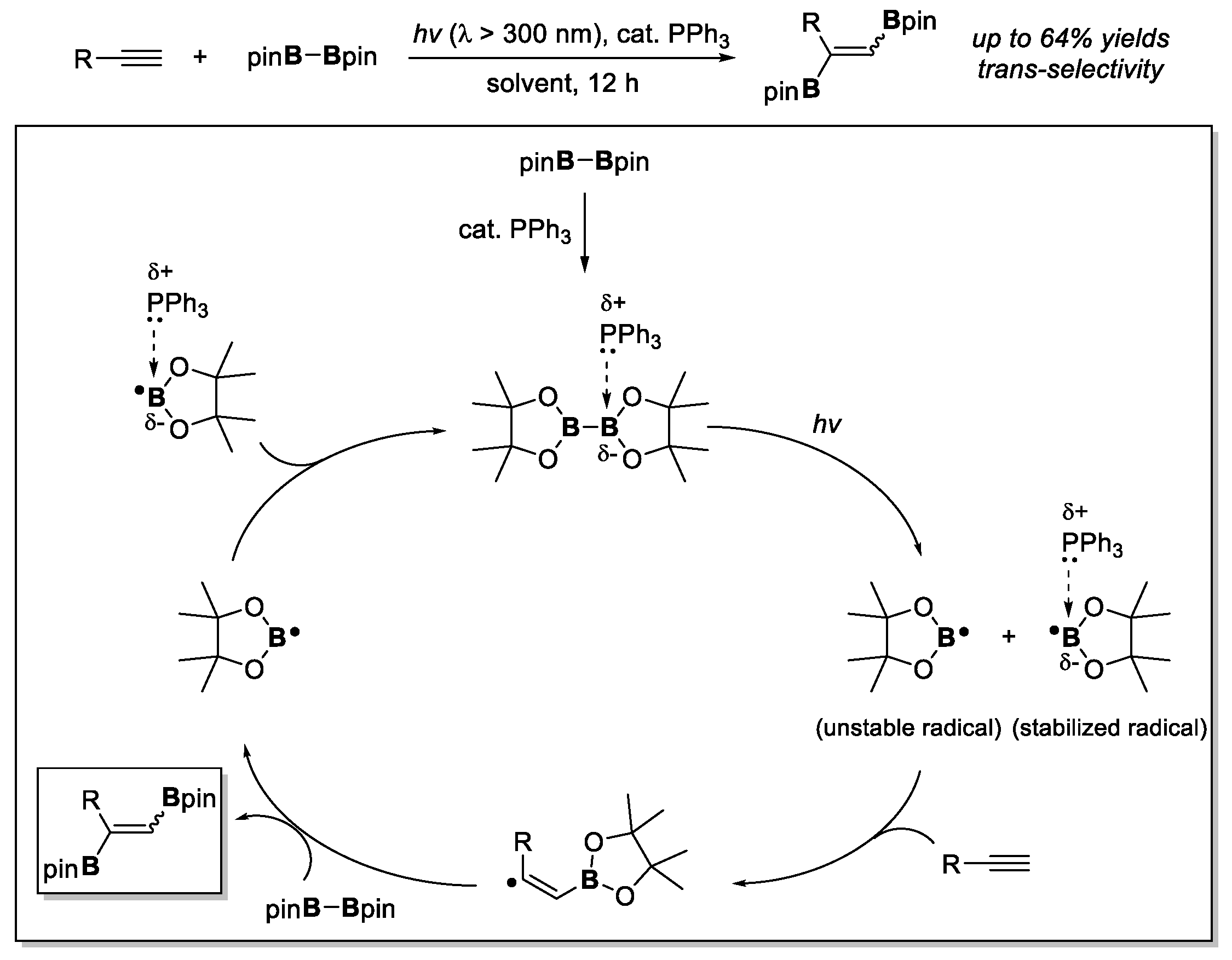
- Yoshimura, A.; Takamachi, Y.; Mihara, K.; Saeki, T.; Kawaguchi, S.-I.; Han, L.-B.; Nomoto, A.; Ogawa, A. Photoinduced metal-free diboration of alkynes in the presence of organophosphine catalysts. Tetrahedron 2016, 72, 7832–7838. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, H.; Zhao, J.; Li, W.; Cao, J.; Zhu, C.; Li, S. Homolytic Cleavage of a B−B Bond by the Cooperative Catalysis of Two Lewis Bases: Computational Design and Experimental Verification. Angew. Chem. Int. Ed. 2016, 55, 5985–5989. [Google Scholar] [CrossRef]
- Verma, P.K.; Meher, N.K.; Geetharani, K. Homolytic cleavage of diboron(4) compounds using diazabutadiene derivatives. Chem. Commun. 2021, 57, 7886–7889. [Google Scholar] [CrossRef]
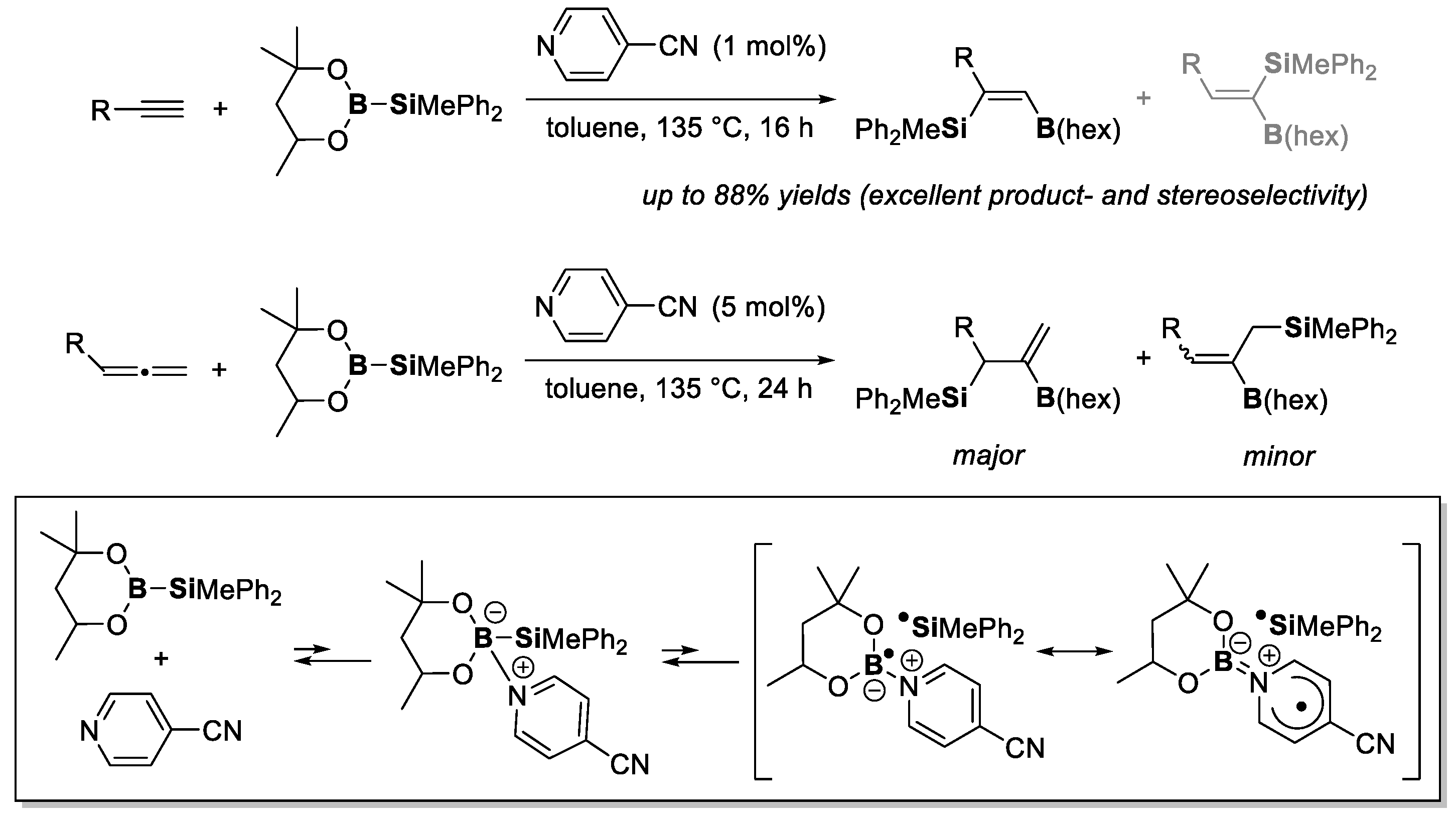
- Tsurugi, H.; Mashima, K.; Castro, L.C.M.; Sultan, I. Pyridine-Mediated B–B Bond Activation of (RO)2B–B(OR)2 for Generating Borylpyridine Anions and Pyridine-Stabilized Boryl Radicals as Useful Boryl Reagents in Organic Synthesis. Synthesis 2021, 53, 3211–3226. [Google Scholar] [CrossRef]
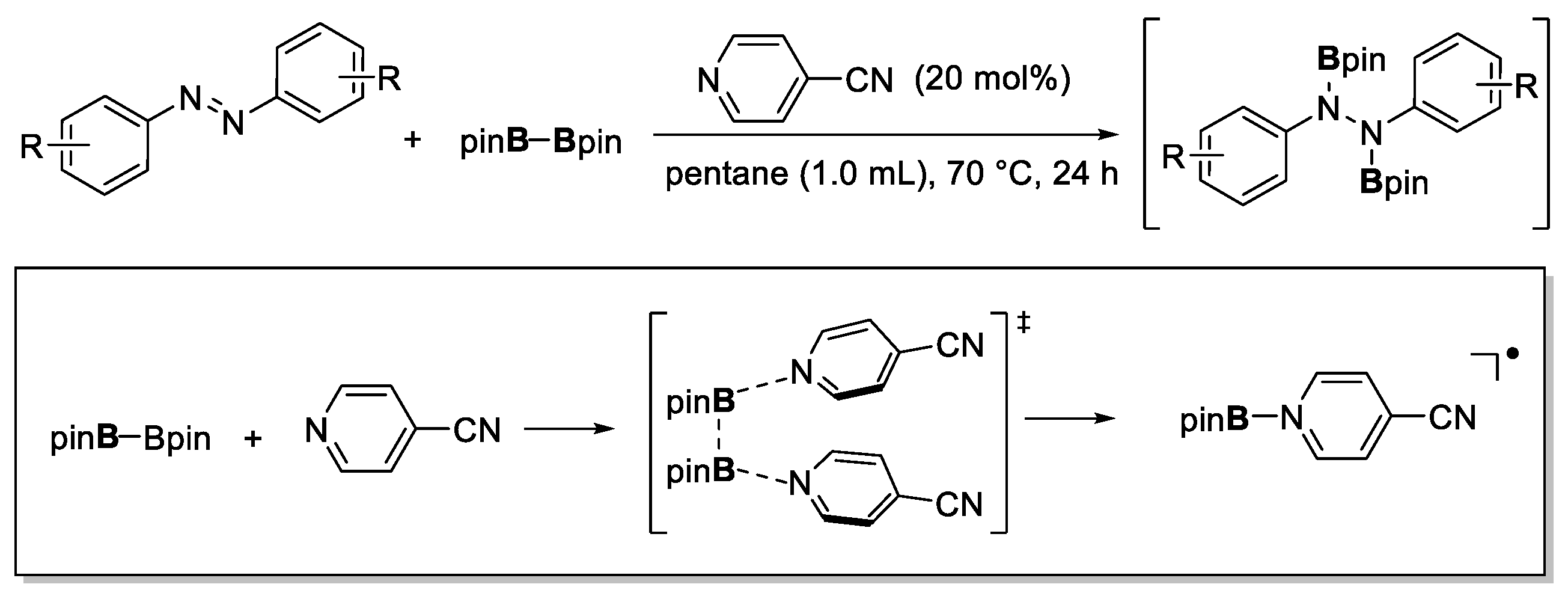
- Xu, R.; Lu, G.-P.; Cai, C. 4-Cyanopyridine-catalyzed anti-Markovnikov selective hydroboration of alkenes. New J. Chem. 2018, 42, 16456–16459. [Google Scholar] [CrossRef]
- Morimasa, Y.; Kabasawa, K.; Ohmura, T.; Suginome, M. Pyridine-Based Organocatalysts for Regioselective syn-1,2-Silaboration of Terminal Alkynes and Allenes. Asian J. Org. Chem. 2019, 8, 1092–1096. [Google Scholar] [CrossRef]
- Gao, L.; Liu, X.; Li, G.; Chen, S.; Cao, J.; Wang, G.; Li, S. 1,2-Silylpyridylation Reaction of Aryl Alkenes with Silylboronate. Org. Lett. 2022, 24, 5698–5703. [Google Scholar] [CrossRef]
- Cao, J.; Li, G.; Wang, G.; Gao, L.; Li, S. Iodoperfluoroalkylation of unactivated alkenes via pyridine-boryl radical initiated atom-transfer radical addition. Org. Biomol. Chem. 2022, 20, 2857–2862. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mück-Lichtenfeld, C.; Studer, A. 1,n-Bisborylalkanes via Radical Boron Migration. J. Am. Chem. Soc. 2020, 142, 9119–9123. [Google Scholar] [CrossRef]
- Cheng, Y.; Mück-Lichtenfeld, C.; Studer, A. Transition Metal-Free 1,2-Carboboration of Unactivated Alkenes. J. Am. Chem. Soc. 2018, 140, 6221–6225. [Google Scholar] [CrossRef]
- Ueng, S.-H.; Solovyev, A.; Yuan, X.; Geib, S.J.; Fensterbank, L.; Lacôte, E.; Malacria, M.; Newcomb, M.; Walton, J.C.; Curan, D.P. N-Heterocyclic Carbene Boryl Radicals: A New Class of Boron-Centered Radical. J. Am. Chem. Soc. 2009, 131, 11256–11262. [Google Scholar] [CrossRef]
- Curran, D.P.; Solovyev, A.; Brahmi, M.M.; Fensterbank, L.; Malacria, M.; Lacôte, E. Synthesis and Reactions of N-Heterocyclic. Carbene Boranes. Angew. Chem. Int. Ed. 2011, 50, 10294–10317. [Google Scholar] [CrossRef]
- Kawamoto, T.; Geib, S.J.; Curran, D.P. Radical Reactions of N-Heterocyclic Carbene Boranes with Organic Nitriles: Cyanation. of NHC-Boranes and Reductive Decyanation of Malononitriles. J. Am. Chem. Soc. 2015, 137, 8617–8622. [Google Scholar] [CrossRef]
- Studer, A.; Curran, D.P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed. 2016, 55, 58–102. [Google Scholar] [CrossRef]
- Kuehn, L.; Zapf, L.; Werner, L.; Stang, M.; Würtemberger-Pietsch, S.; Krummenacher, I.; Braunschweig, H.; Lacôte, E.; Marder, T.B.; Radius, U. NHC induced radical formation via homolytic cleavage of B–B bonds and its role in organic reactions. Chem. Sci. 2022, 13, 8321–8333. [Google Scholar] [CrossRef] [PubMed]
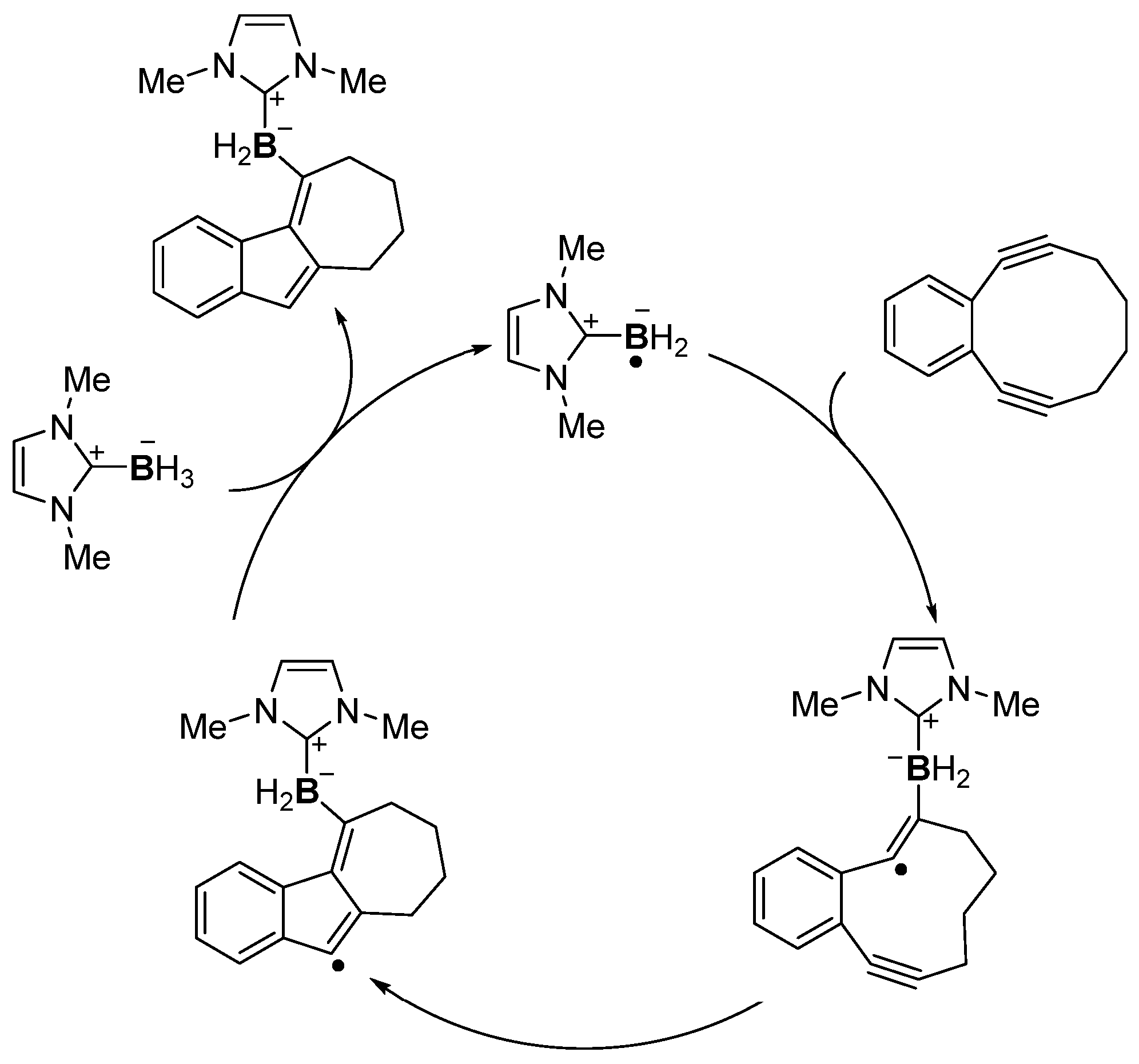
- Watanabe, T.; Hirose, D.; Curran, D.P.; Taniguchi, T. Borylative Radical Cyclizations of Benzo[3,4]cyclodec-3-ene-1,5-diynes and N-Heterocyclic Carbene-Boranes. Chem. Eur. J. 2017, 23, 5404–5409. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.-C.; Zhang, F.-L.; Qi, J.; Huang, Y.-S.; Xu, A.-Q.; Yan, H.-Y.; Wang, Y.-F. Radical Borylation/Cyclization Cascade of 1,6-Enynes for the Synthesis of Boron-Handled Hetero- and Carbocycles. J. Am. Chem. Soc. 2017, 139, 6050–6053. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shen, Y.; Lu, C.; Jian, Y.; Xia, S.; Gao, Z.; Zheng, Y.; An, Y.; Wang, Y. Visible-light-driven PhSSPh-catalysed regioselective hydroborylation of α,β-unsaturated carbonyl compounds with NHC-boranes. Chem. Commun. 2022, 58, 8380–8383. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Nishimoto, Y.; Yasuda, M. (o-Phenylenediamino)borylstannanes: Efficient Reagents for Borylation of Various Alkyl Radical Precursors. Chem. A Eur. J. 2021, 27, 3968–3973. [Google Scholar] [CrossRef]
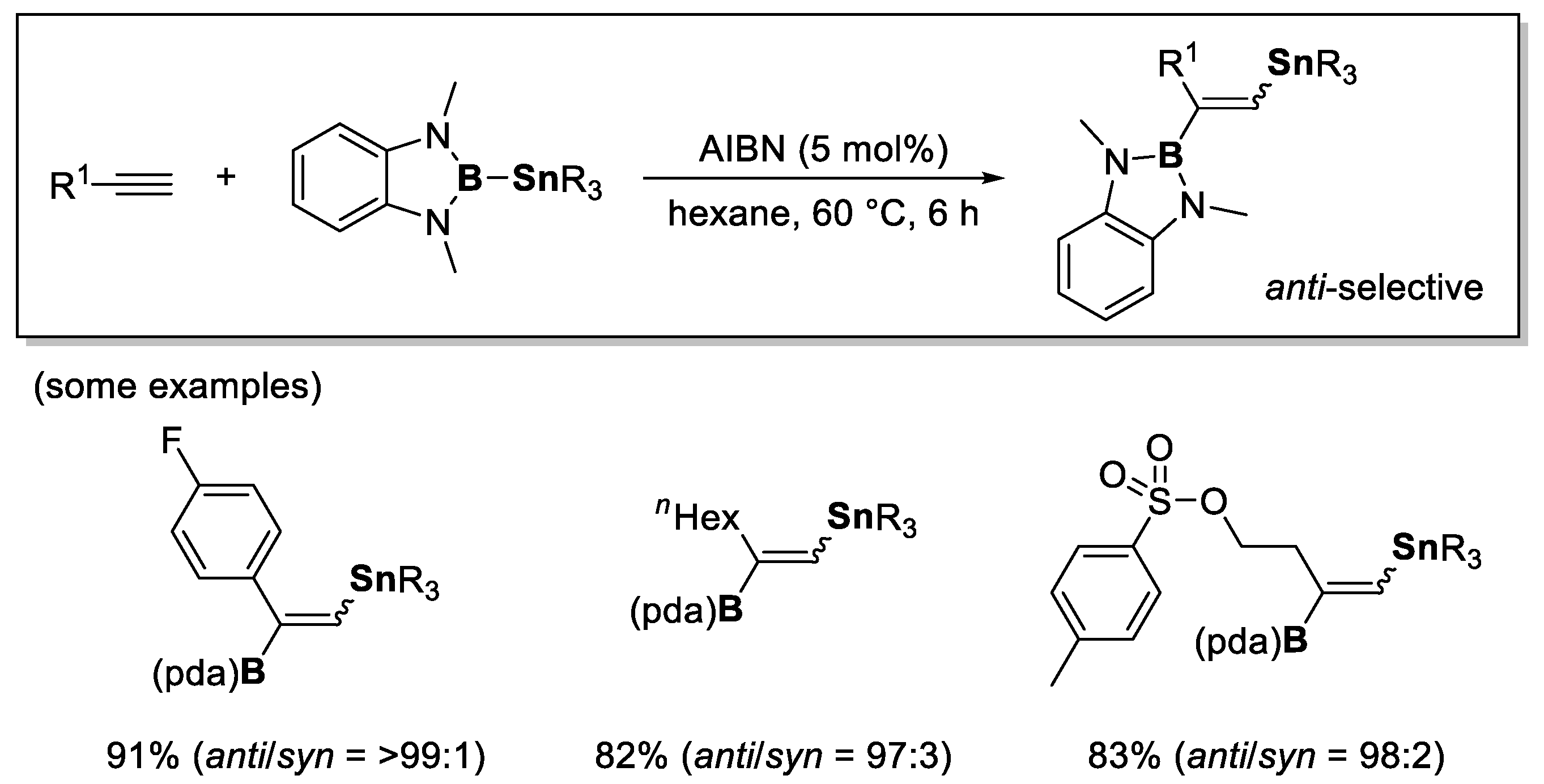
- Suzuki, K.; Sugihara, N.; Nishimoto, Y.; Yasuda, M. anti-Selective Borylstannylation of Alkynes with (o-Phenylenediaminato)borylstannanes by a Radical Mechanism. Angew. Chem. Int. Ed. 2022, 61, e202201883. [Google Scholar] [CrossRef]





















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, Y.; Ogawa, A. Metal-Free One-Pot Multi-Functionalization of Unsaturated Compounds with Interelement Compounds by Radical Process. Molecules 2023, 28, 787. https://doi.org/10.3390/molecules28020787
Yamamoto Y, Ogawa A. Metal-Free One-Pot Multi-Functionalization of Unsaturated Compounds with Interelement Compounds by Radical Process. Molecules. 2023; 28(2):787. https://doi.org/10.3390/molecules28020787
Chicago/Turabian StyleYamamoto, Yuki, and Akiya Ogawa. 2023. "Metal-Free One-Pot Multi-Functionalization of Unsaturated Compounds with Interelement Compounds by Radical Process" Molecules 28, no. 2: 787. https://doi.org/10.3390/molecules28020787