
The Halogen Bond in Weakly Bonded Complexes and the Consequences for Aromaticity and Spin-Orbit Coupling
 , , ,
, , ,
Abstract
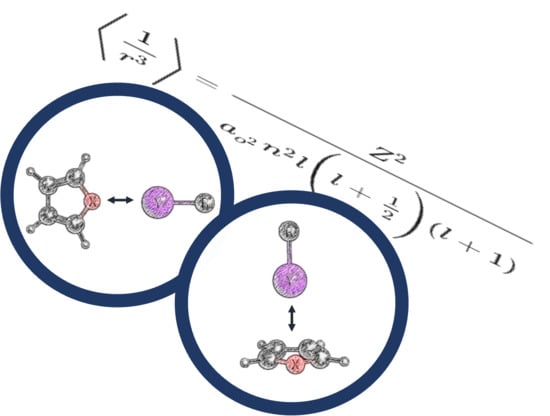
:
1. Introduction
2. Results and Discussion
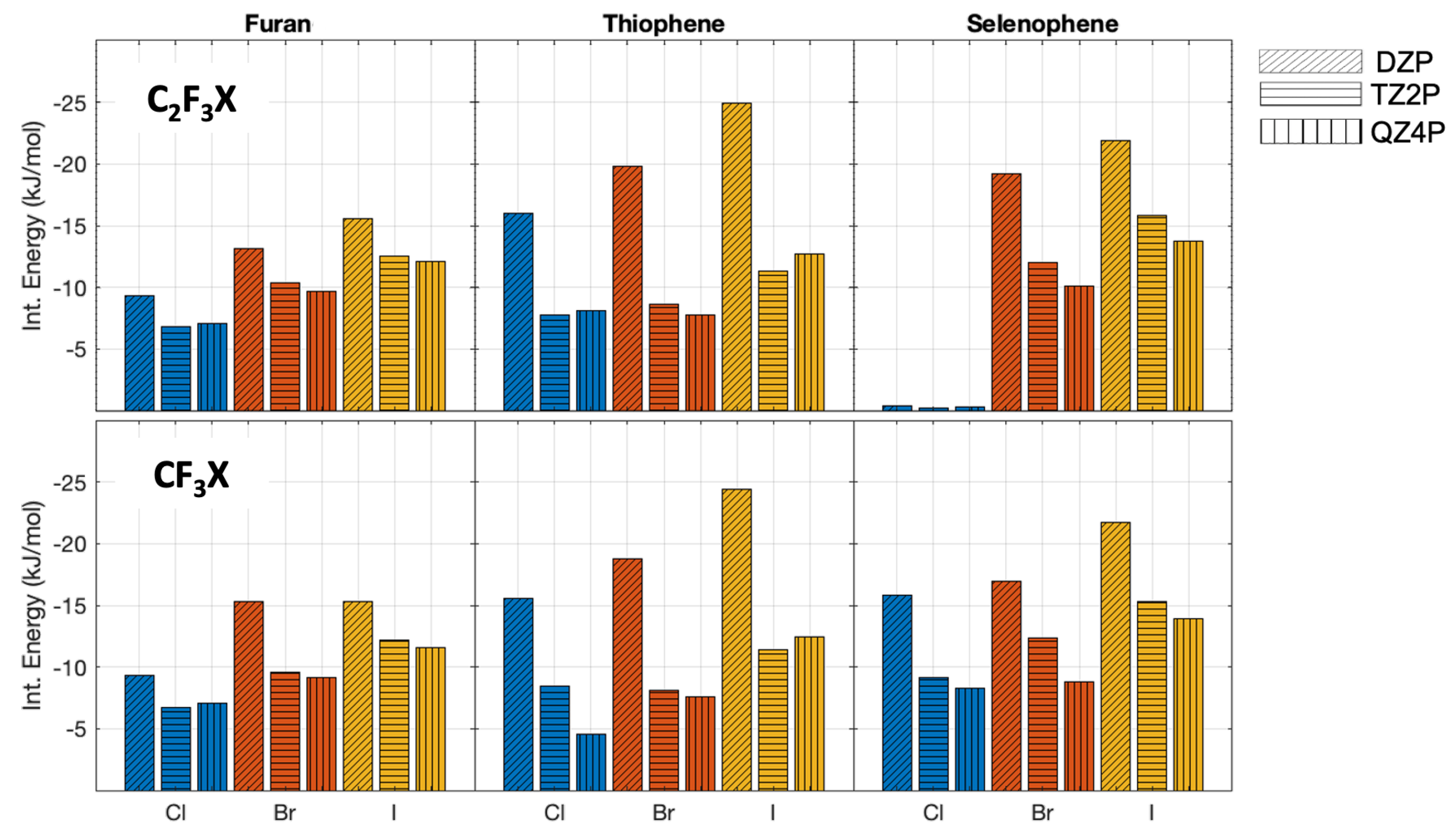
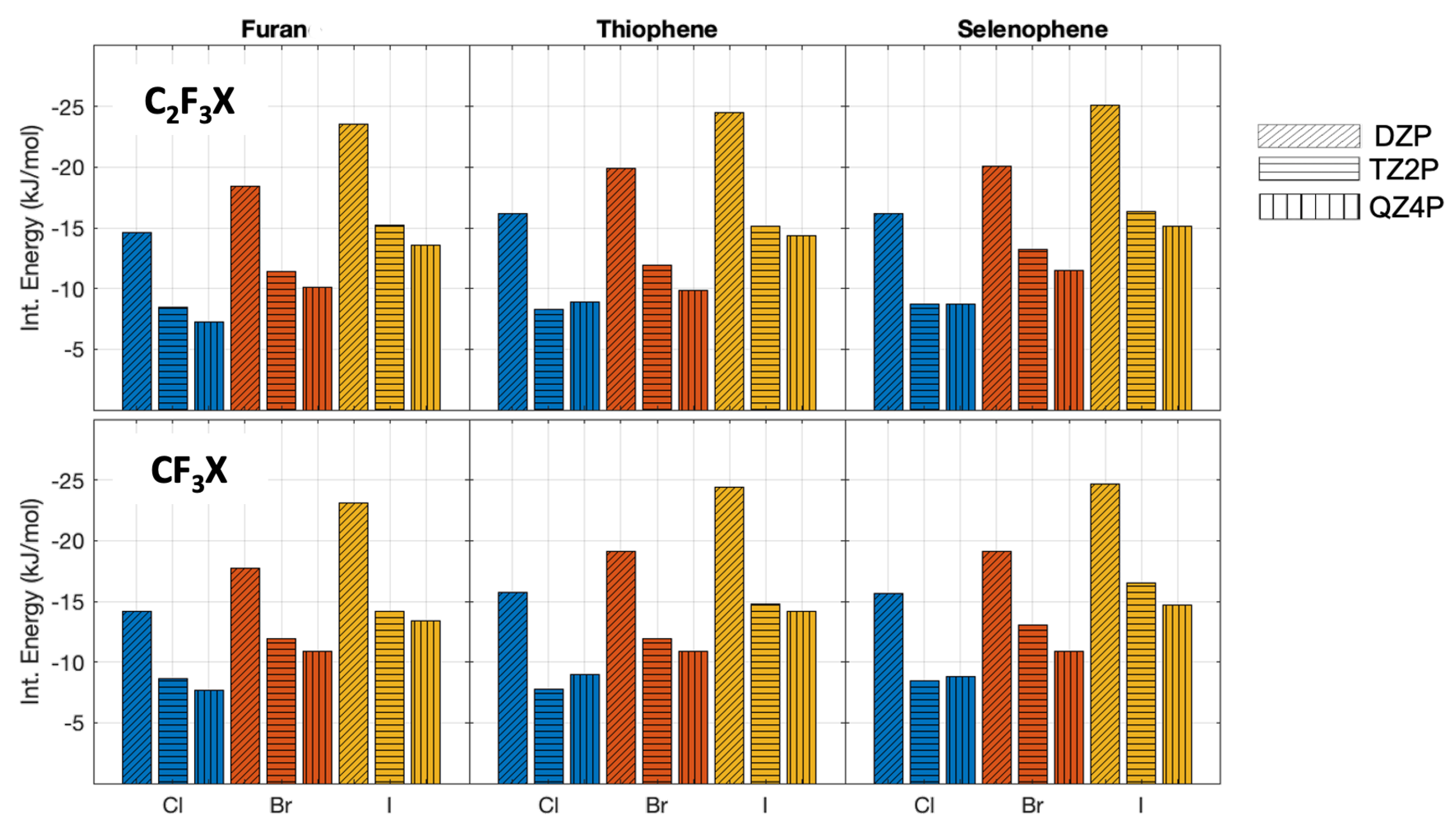
2.1. Interaction Energies at Different Levels of Theory
2.2. Interaction Energies and Trends
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CCSD(T) | Single double coupled cluster with perturbative triples |
| EDA | Energy Decomposition Analysis |
| CDFT | Conceptual Density Functional Theory |
| DFT | Density Functional Theory |
| DZ | Double Zeta |
| TZ2P | Triple-Zeta with two polarization functions |
| QZ4P | Valence Quadruple-Zeta + 4 polarization function, relativistically optimized |
References
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Zingaro, R.A.; Hedges, M. Phosphine Oxide-Halogen Complexes: Effect on P-O and P-S Stretching Frequencies. J. Phys. Chem. 1961, 65, 1132–1138. [Google Scholar] [CrossRef]
- Glaser, R.; Murphy, R.F. What s in a Name? Noncovalent Ar-Cl·(H-Ar)n Interactions and Terminology Based on Structure and Nature of the Bonding. CrystEngComm 2006, 8, 948–951. [Google Scholar] [CrossRef]
- Clark, T.; Murray, J.S.; Lane, P.; Politzer, P. Why Are Dimethyl Sulfoxide and Dimethyl Sulfone Such Good Solvents? J. Mol. Model. 2008, 14, 689–697. [Google Scholar] [CrossRef]
- Bianchi, R.; Forni, A.; Pilati, T. Experimental Electron Density Study of the Supramolecular Aggregation between 4,4-Dipyridyl-N,N′-Dioxide and 1,4-Diiodotetrafluorobenzene at 90 K. Acta Crystallogr. Sect. B Struct. Sci. 2004, 60, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding: An Electrostatically-Driven Highly Directional Noncovalent Interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen Bonding and Other σ-Hole Interactions: A Perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef]
- Hassel, O.; Hvoslef, J. The Structure of Bromine 1,4-Dioxanate. Acta Chem. Scand. 1954, 8, 873. [Google Scholar] [CrossRef] [Green Version]
- Forni, A.; Metrangolo, P.; Pilati, T.; Resnati, G. Halogen Bond Distance as a Function of Temperature. Cryst. Growth Des. 2004, 4, 291–295. [Google Scholar] [CrossRef]
- Khavasi, H.R.; Tehrani, A.A. Influence of Halogen Bonding Interaction on Supramolecular Assembly of Coordination Compounds; Head-to-Tail N⋯X Synthon Repetitivity. Inorg. Chem. 2013, 52, 2891–2905. [Google Scholar] [CrossRef]
- Crihfield, A.; Hartwell, J.; Phelps, D.; Walsh, R.B.; Harris, J.L.; Payne, J.F.; Pennington, W.T.; Hanks, T.W. Crystal Engineering through Halogen Bonding. 2. Complexes of Diacetylene-Linked Heterocycles with Organic Iodides. Cryst. Growth Des. 2003, 3, 313–320. [Google Scholar] [CrossRef]
- Bjorvatten, T.; Hassel, O. Crystal Structure of the 1:3 Addition Compound Iodoform-Quinoline. Acta Chem. Scand. 1962, 16, 249–255. [Google Scholar] [CrossRef]
- Pigge, F.C.; Vangala, V.R.; Swenson, D.C. Relative Importance of X … O=C vs. X … X Halogen Bonding as Structural Determinants in 4-Halotriaroylbenzenes. Chem. Commun. 2006, 2123–2125. [Google Scholar] [CrossRef] [PubMed]
- Evangelisti, L.; Feng, G.; Gou, Q.; Grabow, J.U.; Caminati, W. Halogen Bond and Free Internal Rotation: The Microwave Spectrum of CF3Cl-Dimethyl Ether. J. Phys. Chem. A 2014, 118, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Syssa-Magalé, J.L.; Boubekeur, K.; Palvadeau, P.; Meerschaut, A.; Schöllhorn, B. Self-Assembly via (N⋯I) Non-Covalent Bonds between 1,4-Diiodo-Tetrafluoro-Benzene and a Tetra-Imino Ferrocenophane. J. Mol. Struct. 2004, 691, 79. [Google Scholar] [CrossRef]
- Holmesland, O.; Romming, C. Crystal Structure of the (1:1) Addition Compounds of Diiodoacetylene with 1,4-Dithiane and 1,4-Dieselenane Respectively. Acta Chem. Scand. 1966, 20, 2601. [Google Scholar] [CrossRef]
- Cinčić, D.; Friščić, T.; Jones, W. Experimental and Database Studies of Three-Centered Halogen Bonds with Bifurcated Acceptors Present in Molecular Crystals, Cocrystals and Salts. CrystEngComm 2011, 13, 3224–3231. [Google Scholar] [CrossRef]
- Raatikainen, K.; Huuskonen, J.; Lahtinen, M.; Metrangolo, P.; Rissanen, K. Halogen Bonding Drives the Self-Assembly of Piperazine Cyclophanes into Tubular Structures. Chem. Commun. 2009, 16, 2160–2162. [Google Scholar] [CrossRef]
- Nagels, N.; Geboes, Y.; Pinter, B.; De Proft, F.; Herrebout, W.A. Tuning the Halogen/Hydrogen Bond Competition: A Spectroscopic and Conceptual DFT Study of Some Model Complexes Involving CHF2I. Chem. Eur. J. 2014, 20, 8433–8443. [Google Scholar] [CrossRef]
- Herrebout, W. Infrared and Raman Measurements of Halogen Bonding in Cryogenic Solutions; Springer International Publishing: Cham, Switzerland, 2015; pp. 79–154. [Google Scholar]
- Thirman, J.; Engelage, E.; Huber, S.M.; Head-Gordon, M. Characterizing the interplay of Pauli repulsion, electrostatics, dispersion and charge transfer in halogen bonding with energy decomposition analysis. Phys. Chem. Chem. Phys. 2018, 20, 905–915. [Google Scholar] [CrossRef]
- Messina, M.T.; Metrangolo, P.; Panzeri, W.; Ragg, E.; Resnati, G. Perfluorocarbon-Hydrocarbon Self-Assembly. Part 3. Liquid Phase Interactions between Perfluoroalkylhalides and Heteroatom Containing Hydrocarbons. Tetrahedron Lett. 1998, 39, 9069–9072. [Google Scholar] [CrossRef]
- Metrangolo, P.; Panzeri, W.; Recupero, F.; Resnati, G. Perfluorocarbon-Hydrocarbon Self-Assembly Part 16. 19F NMR Study of the Halogen Bonding between Halo-Perfluorocarbons and Heteroatom Containing Hydrocarbons. J. Fluorine Chem. 2002, 114, 27–33. [Google Scholar] [CrossRef]
- Bjorvatten, T. Crystal Structures of Chloro and Bromo Cyanoacetylene. Acta Chem. Scand. 1968, 22, 410. [Google Scholar] [CrossRef] [Green Version]
- Geboes, Y.; Nagels, N.; Pinter, B.; De Proft, F.; Herrebout, W.A. Competition of C(sp2)-X⋯O Halogen Bonding and Lone Pair⋯π Interactions: Cryospectroscopic Study of the Complexes of C2F3X (X = F, Cl, Br, and I) and Dimethyl Ether. J. Phys. Chem. A 2015, 119, 2502–2516. [Google Scholar] [CrossRef]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. The Fluorine Atom as a Halogen Bond Donor, Viz. a Positive Site. CrystEngComm 2011, 13, 6593–6596. [Google Scholar] [CrossRef] [Green Version]
- Metrangolo, P.; Murray, J.S.; Pilati, T.; Politzer, P.; Resnati, G.; Terraneo, G. Fluorine-Centered Halogen Bonding: A Factor in Recognition Phenomena and Reactivity. Cryst. Growth Des. 2011, 11, 4238–4246. [Google Scholar] [CrossRef]
- Rege, P.D.; Malkina, O.L.; Goroff, N.S. The Effect of Lewis Bases on the 13C NMR of Iodoalkynes. J. Am. Chem. Soc. 2002, 124, 370–371. [Google Scholar] [CrossRef]
- Perkins, C.; Libri, S.; Adams, H.; Brammer, L. Diiodoacetylene: Compact, Strong Ditopic Halogen Bond Donor. CrystEngComm 2012, 14, 3033–3038. [Google Scholar] [CrossRef]
- Bock, H.; Holl, S. Crystallization and Structure Determination of σ-Donor-Acceptor Complexes between 1,4-Dioxane and the Polyiodine Molecules I2, I2C=CI2, (IC)4S and (IC)4NR. Z. Naturforsch. B J. Chem. Sci. 2001, 56, 111–121. [Google Scholar] [CrossRef]
- Gagnaux, P.; Susz, B.P. Etudes de Composés D’addition Des Acides de LEWIS. XII. Structure, Spectre Infrarouge et Polarisation Moléculaire Du Composé D’addition Dioxanne-1, 4–diiodacétylène. Helv. Chim. Acta 1960, 43, 948. [Google Scholar] [CrossRef]
- Glaser, R.; Chen, N.; Wu, H.; Knotts, N.; Kaupp, M. 13C NMR Study of Halogen Bonding of Haloarenes: Measurements of Solvent Effects and Theoretical Analysis. J. Am. Chem. Soc. 2004, 126, 4412–4419. [Google Scholar] [CrossRef] [PubMed]
- Raatikainen, K.; Cametti, M.; Rissanen, K. The Subtle Balance of Weak Supramolecular Interactions: The Hierarchy of Halogen and Hydrogen Bonds in Haloanilinium and Halopyridinium Salts. Beilstein J. Org. Chem. 2010, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logothetis, T.A.; Meyer, F.; Metrangolo, P.; Pilati, T.; Resnati, G. Crystal Engineering of Brominated Tectons: N-Methyl-3,5-Dibromo-Pyridinium Iodide Gives Particularly Short C-Br⋯I Halogen Bonding. New J. Chem. 2004, 28, 760. [Google Scholar] [CrossRef]
- Liantonio, R.; Metrangolo, P.; Pilati, T.; Resnati, G. Fluorous Interpenetrated Layers in a Three-Component Crystal Matrix. Cryst. Growth Des. 2003, 3, 355–361. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Fasulo, M.; Schultheiss, N.; Desper, J.; Moore, C. Structural Competition between Hydrogen Bonds and Halogen Bonds. J. Am. Chem. Soc. 2007, 129, 13772–13773. [Google Scholar] [CrossRef]
- Saccone, M.; Cavallo, G.; Metrangolo, P.; Pace, A.; Pibiri, I.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bond Directionality Translates Tecton Geometry into Self-Assembled Architecture Geometry. CrystEngComm 2013, 15, 3102–3105. [Google Scholar] [CrossRef] [Green Version]
- Messina, M.T.; Metrangolo, P.; Panzeri, W.; Pilati, T.; Resnati, G. Intermolecular Recognition between Hydrocarbon Oxygen-Donors and Perfluorocarbon Iodine-Acceptors: The Shortest O⋯I Non-Covalent Bond. Tetrahedron 2001, 57, 8543. [Google Scholar] [CrossRef]
- Syssa-Magalé, J.L.; Boubekeur, K.; Leroy, J.; Chamoreau, L.M.; Fave, C.; Schöllhorn, B. Directed Synthesis of a Halogen-Bonded Open Porphyrin Network. CrystEngComm 2014, 16, 10380–10384. [Google Scholar] [CrossRef]
- Syssa-Magalé, J.L.; Boubekeur, K.; Schöllhorn, B. First Molecular Self-Assembly of 1,4-Diiodo-Tetrafluoro-Benzene and a Ketone via (O⋯I) Non-Covalent Halogen Bonds. J. Mol. Struct. 2005, 737, 103. [Google Scholar] [CrossRef]
- Parisini, E.; Metrangolo, P.; Pilati, T.; Resnati, G.; Terraneo, G. Halogen Bonding in Halocarbon-Protein Complexes: A Structural Survey. Chem. Soc. Rev. 2011, 40, 2267–2278. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen Bonds in Biological Molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, S.S.; Praveen, V.K.; Ajayaghosh, A. Functional π-Gelators and Their Applications. Chem. Rev. 2014, 114, 1973–2129. [Google Scholar]
- Bartelena, L.; Robbins, J. Thyroid Hormone Transport Proteins. Clin. Lab. Med. 1993, 13, 583–598. [Google Scholar] [CrossRef]
- Xu, Z.; Yang, Z.; Liu, Y.; Lu, Y.; Chen, K.; Zhu, W. Halogen Bond: Its Role beyond Drug-Target Binding Affinity for Drug Discovery and Development. J. Chem. Inf. Model. 2014, 54, 69–78. [Google Scholar] [CrossRef]
- Howard, E.I.; Sanishvili, R.; Cachau, R.E.; Mitschler, A.; Chevrier, B.; Barth, P.; Lamour, V.; Van Zandt, M.; Sibley, E.; Bon, C. Ultrahigh Resolution Drug Design I: Details of Interactions in Human Aldose Reductase-Inhibitor Complex at 0.66 Å. Proteins Struct. Funct. Genet. 2004, 55, 792–804. [Google Scholar] [CrossRef]
- Hays, F.A.; Vargason, J.M.; Ho, P.S. Effect of Sequence on the Conformation of DNA Holliday Junctions. Biochemistry 2003, 42, 9586–9597. [Google Scholar] [CrossRef] [Green Version]
- El-Kabbani, O.; Ramsland, P.; Darmanin, C.; Chung, R.P.T.; Podjarny, A. Structure of Human Aldose Reductase Holoenzyme in Complex with Statil: An Approach to Structure-Based Inhibitor Design of the Enzyme. Proteins Struct. Funct. Genet. 2003, 50, 230–238. [Google Scholar] [CrossRef]
- Hays, F.A.; Teegarden, A.; Jones, Z.J.R.; Harms, M.; Raup, D.; Watson, J.; Cavaliere, E.; Ho, P.S. How Sequence Defines Structure: A Crystallographic Map of DNA Structure and Conformation. Proc. Natl. Acad. Sci. USA 2005, 102, 7157–7162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichman, B.F.; Vargason, J.M.; Mooers, B.H.M.; Ho, P.S. The Holliday Junction in an Inverted Repeat DNA Sequence: Sequence Effects on the Structure of Four-Way Junctions. Proc. Natl. Acad. Sci. USA 2000, 97, 3971–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolton, O.; Lee, K.; Kim, H.J.; Lin, K.Y.; Kim, J. Activating Efficient Phosphorescence from Purely Organic Materials by Crystal Design. Nat. Chem. 2011, 3, 205–210. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Park, S.Y. Phosphorescent iridium(III) Complexes: Toward High Phosphorescence Quantum Efficiency through Ligand Control. Dalton Trans. 2009, 8, 1267–1282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Huang, C.; Li, F. Phosphorescent Heavy-Metal Complexes for Bioimaging. Chem. Soc. Rev. 2011, 40, 2508–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, W.Z.; Shen, X.Y.; Zhao, H.; Lam, J.W.Y.; Tang, L.; Lu, P.; Wang, C.; Liu, Y.; Wang, Z.; Zheng, Q. Crystallization-Induced Phosphorescence of Pure Organic Luminogens at Room Temperature. J. Phys. Chem. C 2010, 114, 6090–6099. [Google Scholar] [CrossRef]
- Bolton, O.; Lee, D.; Jung, J.; Kim, J. Tuning the Photophysical Properties of Metal-Free Room Temperature Organic Phosphors via Compositional Variations in Bromobenzaldehyde/Dibromobenzene Mixed Crystals. Chem. Mater. 2014, 26, 6644–6649. [Google Scholar] [CrossRef]
- Kwon, M.S.; Lee, D.; Seo, S.; Jung, J.; Kim, J. Tailoring Intermolecular Interactions for Efficient Room-Temperature Phosphorescence from Purely Organic Materials in Amorphous Polymer Matrices. Angew. Chem. Int. Ed. 2014, 53, 11177. [Google Scholar] [CrossRef]
- Gao, H.Y.; Zhao, X.R.; Wang, H.; Pang, X.; Jin, W.J. Phosphorescent Cocrystals Assembled by 1,4- Diiodotetrafluorobenzene and Fluorene and Its Heterocyclic Analogues Based on C–I⋯π Halogen Bonding. Cryst. Growth Des. 2012, 12, 4377–4387. [Google Scholar] [CrossRef]
- Shi, L.; Liu, H.Y.; Shen, H.; Hu, J.; Zhang, G.L.; Wang, H.; Ji, L.N.; Chang, C.K.; Jiang, H.F. Fluorescence Properties of Halogenated Mono-Hydroxyl Corroles: The Heavy-Atom Effects. J. Porphyr. Phthalocyanines 2009, 13, 1221–1226. [Google Scholar] [CrossRef]
- Hirata, S.; Totani, K.; Zhang, J.; Yamashita, T.; Kaji, H.; Marder, S.R.; Watanabe, T.; Adachi, C. Efficient Persistent Room Temperature Phosphorescence in Organic Amorphous Materials under Ambient Conditions. Adv. Funct. Mater. 2013, 23, 3386–3397. [Google Scholar] [CrossRef]
- Lee, D.; Bolton, O.; Kim, B.C.; Youk, J.H.; Takayama, S.; Kim, J. Room Temperature Phosphorescence of Metal-Free Organic Materials in Amorphous Polymer Matrices. J. Am. Chem. Soc. 2013, 135, 6325–6329. [Google Scholar] [CrossRef]
- Pinter, B.; Nagels, N.; Herrebout, W.A.; De Proft, F. Halogen Bonding from a Hard and Soft Acids and Bases Perspective: Investigation by Using Density Functional Theory Reactivity Indices. Chem. Eur. J. 2013, 19, 519–530. [Google Scholar] [CrossRef]
- Gao, H.Y.; Shen, Q.J.; Zhao, X.R.; Yan, X.Q.; Pang, X.; Jin, W.J. Phosphorescent Co-Crystal Assembled by 1,4-Diiodotetrafluorobenzene with Carbazole Based on C–I⋯π Halogen Bonding. J. Mater. Chem. 2012, 22, 5336. [Google Scholar] [CrossRef]
- Zhu, Q.; Gao, Y.J.; Gao, H.Y.; Jin, W.J. Effect of N-Methyl and Ethyl on Phosphorescence of Carbazole in Cocrystals Assembled by C-I⋯π Halogen Bond, π-Hole⋯π Bond and Other Interactions Using 1,4-Diiodotetrafluorobenzene as Donor. J. Photochem. Photobiol. A 2014, 289, 31. [Google Scholar] [CrossRef]
- Shen, Q.J.; Wei, H.Q.; Zou, W.S.; Sun, H.L.; Jin, W.J. Cocrystals Assembled by Pyrene and 1,2- or 1,4-Diiodotetrafluorobenzenes and Their Phosphorescent Behaviors Modulated by Local Molecular Environment. CrystEngComm 2012, 14, 1010–1015. [Google Scholar] [CrossRef]
- Pang, X.; Wang, H.; Zhao, X.R.; Jin, W.J. Co-Crystallization Turned on the Phosphorescence of Phenanthrene by C–Br⋯π Halogen Bonding, π–hole⋯π Bonding and Other Assisting Interactions. CrystEngComm 2013, 15, 2722–2730. [Google Scholar] [CrossRef]
- Zhu, Q.; Wang, H.; Zhao, X.R.; Jin, W.J. The Phosphorescent Behaviors of 9-Bromo- and 9-Iodophenanthrene in Crystals Modulated by π-π Interactions, C-H⋯π Hydrogen Bond and C-I⋯π Halogen Bond. J. Photochem. Photobiol. A 2014, 274, 98–107. [Google Scholar] [CrossRef]
- Stöhr, M.; Van Voorhis, T.; Tkatchenko, A. Theory and practice of modeling van der Waals interactions in electronic-structure calculations. Chem. Soc. Rev. 2019, 48, 4118–4154. [Google Scholar] [CrossRef] [Green Version]
- Otero-de-la Roza, A.; Johnson, E.R. Van der Waals interactions in solids using the exchange-hole dipole moment model. J. Chem. Phys. 2012, 136, 174109. [Google Scholar] [CrossRef]
- Otero-de-la Roza, A.; Johnson, E.R. A benchmark for non-covalent interactions in solids. J. Chem. Phys. 2012, 137, 054103. [Google Scholar] [CrossRef] [Green Version]
- Gobre, V.V.; Tkatchenko, A. Scaling laws for van der Waals interactions in nanostructured materials. Nat. Commun. 2013, 4, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Reilly, A.M.; Tkatchenko, A. van der Waals dispersion interactions in molecular materials: Beyond pairwise additivity. Chem. Sci 2015, 6, 3289–3301. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Klimes, J.; Michaelides, A. Perspective: Advances and challenges in treating van der Waals dispersion forces in density functional theory. J. Chem. Phys. 2012, 137, 120901. [Google Scholar] [CrossRef] [Green Version]
- Bjorkman, T. Testing several recent van der Waals density functionals for layered structures. J. Chem. Phys. 2014, 141, 074708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosetti, A.; Reilly, A.M.; DiStasio, R.A.; Tkatchenko, A. Long-range correlation energy calculated from coupled atomic response functions. J. Chem. Phys. 2014, 140, 18A508. [Google Scholar] [CrossRef] [Green Version]
- Sinanoglu, O. Many-Electron Theory of Atoms, Molecules and their interactions. Adv. Chem. Phys. 1964, 6, 315–412. [Google Scholar]
- Buhmann, S.Y. Dispersion Forces I; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Woods, L.M.; Dalvit, D.A.R.; Tkatchenko, A.; Rodriguez-Lopez, P.; Rodriguez, A.W.; Podgornik, R. Materials perspective on Casimir and van der Waals interactions. Rev. Mod. Phys. 2016, 88, 045003. [Google Scholar] [CrossRef] [Green Version]
- Dobson, J.F.; White, A.; Rubio, A. Asymptotics of the dispersion interaction: Analytic benchmarks for van der Waals energy functionals. Phys. Rev. Lett. 2006, 96, 073201. [Google Scholar] [CrossRef] [Green Version]
- Blum, V.; Gehrke, R.; Hanke, F.; Havu, P.; Havu, V.; Ren, X.G.; Reuter, K.; Scheffler, M. Ab initio molecular simulations with numeric atom-centered orbitals. Comp. Phys. Commun. 2009, 180, 2175–2196. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef] [Green Version]
- Jiménez-Grávalos, F.; Gallegos, M.; Martín Pendás, A.; Novikov, A.S. Challenging the electrostatic σ-hole picture of halogen bonding using minimal models and the interacting quantum atoms approach. J. Comput. Chem. 2021, 42, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Steiner, E.; Fowler, P.W. Patterns of ring currents in conjugated molecules: A few-electron model based on orbital contributions. J. Phys. Chem. A 2001, 105, 9553–9562. [Google Scholar] [CrossRef]
- Steiner, E.; Fowler, P.W. Four- and two-electron rules for diatropic and paratropic ring currents in monocyclic π systems. Chem. Commun. 2001, 21, 2220–2221. [Google Scholar] [CrossRef]
- De Vleeschouwer, F.; De Proft, F.; Ergün, O.; Herrebout, W.; Geerlings, P. A Combined Experimental/Quantum-Chemical Study of Tetrel, Pnictogen, and Chalcogen Bonds of Linear Triatomic Molecules. Molecules 2021, 26, 6767. [Google Scholar] [CrossRef]
- Steinmann, S.; Corminboeuf, C. Comprehensive Benchmarking of a Density-Dependent Dispersion Correction. J. Chem. Theory Comput. 2011, 7, 3567–3577. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- van Lenthe, E.; Baerends, E.J.S.J.G. Relativistic total energy using regular approximations. J. Chem. Phys. 1994, 101, 9783–9792. [Google Scholar] [CrossRef]
- van Lenthe, E.; van Leeuwen, R.; Baerends, E.J.; Snijders, J.G. Relativistic regular two-component Hamiltonians. Int. J. Quantum Chem. 1996, 57, 281–293. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M.; Baerends, E.J. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistr. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; Wiley-VCH: New York, NY, USA, 2000; pp. 1–86. [Google Scholar]
- te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Baerends, E.J. AMS2022.01, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands. Available online: http://www.scm.com (accessed on 30 July 2022).
- Liakos, D.; Guo, Y.; Neese, F. Comprehensive benchmark results for the domain based local pair natural orbital coupled cluster method (DLPNO-CCSD(T)) for closed- and open-shell systems. J. Phys. Chem. A 2020, 124, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system—Version 5.0. WIREs Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Guest, M.F.; Bush, I.J.; van Dam, H.J.J.; Sherwood, P.; Thomas, J.M.H.; van Lenthe, J.H.; Havenith, R.W.A.; Kendrick, J. The GAMESS-UK electronic structure package: Algorithms, developments and applications. Mol. Phys. 2005, 103, 719–747. [Google Scholar] [CrossRef]
- Havenith, R.W.A.; Fowler, P. Ipsocentric ring currents in density functional theory. Chem. Phys. Lett. 2007, 449, 347–353. [Google Scholar] [CrossRef]
- Lazzeretti, P.; Zanasi, R. SYSMO Package; Additional Routines by P. W. Fowler, E. Steiner, R. W. A. Havenith, A. Soncini; University of Modena: Modena, Italy, 1980. [Google Scholar]
- Keith, T.; Bader, R.F.W. Calculation of magnetic response properties using a continuous set of gauge transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- Lazzeretti, P.; Malagoli, M.; Zanasi, R. Electronic current density induced by nuclear magnetic dipoles. Chem. Phys. Lett. 1994, 220, 299–304. [Google Scholar] [CrossRef]
- Coriani, S.; Lazzeretti, P.; Malagoli, M.; Zanasi, R. On CHF calculations of second-order magnetic properties using the method of continuous transformation of origin of the current density. Theor. Chim. Acta 1994, 89, 181–192. [Google Scholar] [CrossRef]
- Wang, F.; Ziegler, T. A simplified relativistic time-dependent density-functional theory formalism for the calculations of excitation energies including spin-orbit coupling effect. J. Chem. Phys. 2005, 123, 154102. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Furan | Thiophene | Selenophene | |||||
|---|---|---|---|---|---|---|---|
| CCSD(T) CBS | DFT QZ4P | CCSD(T) CBS | DFT QZ4P | CCSD(T) CBS | DFT QZ4P | ||
| CFX | Cl | −7.2 | −7.0 | −9.1 | −8.1 | −0.5 | −0.3 |
| Br | −9.4 | −9.7 | −8.5 | −7.7 | −10.8 | −10.1 | |
| I | −13.9 | −12.1 | −16.2 | −12.7 | −17.2 | −13.8 | |
| CFX | Cl | −7.0 | −7.1 | −5.1 | −4.6 | −9.6 | −8.3 |
| Br | −9.1 | −9.1 | −8.0 | −7.6 | −8.7 | −8.8 | |
| I | −13.3 | −11.5 | −15.7 | −12.4 | −16.2 | −13.9 | |
| Furan | Thiophene | Selenophene | |||||
|---|---|---|---|---|---|---|---|
| CCSD(T) CBS | DFT QZ4P | CCSD(T) CBS | DFT QZ4P | CCSD(T) CBS | DFT QZ4P | ||
| CFX | Cl | −7.7 | −7.2 | −10.1 | −8.9 | −10.0 | −8.7 |
| Br | −10.4 | −10.1 | −10.8 | −9.9 | −12.3 | −11.5 | |
| I | −15.0 | −13.6 | −17.7 | −14.4 | −17.8 | −15.1 | |
| CFX | Cl | −7.8 | −7.7 | −10.0 | −9.0 | −9.7 | −8.8 |
| Br | −11.0 | −10.9 | −11.8 | −10.9 | −11.3 | −10.9 | |
| I | −14.5 | −13.4 | −16.6 | −14.2 | −17.5 | −14.7 | |
| Compound | H | H | H |
|---|---|---|---|
| CFI⋯furan (‖) | −12.8 | −14.0 | −14.0(8) |
| CFI⋯furan (⊥) | −12.0 | −10.8 | −14.4(9) |
| Complex | ||||||
| CFCl⋯furan | 7.0 | −7.5 | −0.5 | −3.1 | −3.0 | −6.7 |
| CFBr⋯furan | 10.7 | −11.2 | −0.5 | −5.1 | −3.4 | −9.0 |
| CFI⋯furan | 16.9 | −16.7 | 0.2 | −7.9 | −4.2 | −11.9 |
| CFCl⋯thiophene | 7.0 | −6.5 | 0.5 | −3.5 | −4.8 | −7.8 |
| CFBr⋯thiophene | 10.2 | −8.6 | 1.7 | −5.4 | −3.8 | −7.5 |
| CFI⋯thiophene | 18.6 | −14.0 | 4.6 | −10.8 | −6.1 | −12.3 |
| CFCl⋯selenophene | −0.0 | −0.2 | −0.2 | 0.0 | −0.3 | −0.5 |
| CFBr⋯selenophene | 12.1 | −10.1 | 2.0 | −6.9 | −4.8 | −9.7 |
| CFI⋯selenophene | 23.6 | −17.3 | 6.3 | −13.3 | −6.0 | −13.1 |
| CFCl⋯furan | 7.5 | −7.9 | −0.4 | −3.4 | −2.9 | −6.8 |
| CFBr⋯furan | 9.6 | −10.2 | −0.7 | −4.6 | −3.2 | −8.5 |
| CFI⋯furan | 17.6 | −16.9 | 0.6 | −7.9 | −4.1 | −11.4 |
| CFCl⋯thiophene | 1.1 | −2.5 | −1.4 | −1.2 | −2.1 | −4.7 |
| CFBr⋯thiophene | 10.0 | −8.3 | 1.8 | −5.7 | −3.2 | −7.1 |
| CFI⋯thiophene | 20.6 | −15.1 | 5.6 | −11.7 | −5.8 | −12.0 |
| CFCl⋯selenophene | 7.5 | −6.6 | 0.9 | −4.1 | −4.8 | −8.0 |
| CFBr⋯selenophene | 14.5 | −11.4 | 3.1 | −7.8 | −3.6 | −8.2 |
| CFI⋯selenophene | 28.5 | −20.4 | 8.1 | −15.9 | −5.8 | −13.6 |
| Complex | ||||||
| CFCl⋯furan | 5.6 | −5.6 | 0.0 | −2.9 | −4.0 | −7.0 |
| CFBr⋯furan | 13.1 | −11.3 | 1.8 | −6.4 | −4.7 | −9.4 |
| CFI⋯furan | 22.0 | −17.8 | 4.2 | −11.9 | −5.8 | −13.5 |
| CFCl⋯thiophene | 8.2 | −7.1 | 1.1 | −4.0 | −5.3 | −8.3 |
| CFBr⋯thiophene | 8.7 | −8.3 | 0.5 | −4.8 | −5.0 | −9.3 |
| CFI⋯thiophene | 23.7 | −18.3 | 5.4 | −12.3 | −7.1 | −14.1 |
| CFCl⋯selenophene | 7.7 | −6.9 | 0.7 | −3.8 | −5.2 | −8.3 |
| CFBr⋯selenophene | 14.0 | −11.7 | 2.3 | −7.3 | −5.7 | −10.6 |
| CFI⋯selenophene | 23.5 | −18.6 | 4.9 | −12.7 | −7.0 | −14.9 |
| CFCl⋯furan | 9.2 | −8.0 | 1.2 | −4.4 | −4.1 | −7.2 |
| CFBr⋯furan | 16.4 | −12.8 | 3.6 | −8.4 | −5.0 | −9.8 |
| CFI⋯furan | 24.0 | −18.7 | 5.3 | −13.1 | −5.7 | −13.4 |
| CFCl⋯thiophene | 8.2 | −7.2 | 1.0 | −4.2 | −5.1 | −8.3 |
| CFBr⋯thiophene | 14.4 | −11.6 | 2.8 | −7.4 | −5.4 | −10.0 |
| CFI⋯thiophene | 25.9 | −19.9 | 6.1 | −13.7 | −6.7 | −14.3 |
| CFCl⋯selenophene | 7.6 | −7.1 | 0.5 | −3.9 | −5.0 | −8.3 |
| CFBr⋯selenophene | 16.2 | −12.6 | 3.5 | −8.2 | −5.3 | −9.9 |
| CFI⋯selenophene | 27.6 | −20.6 | 7.1 | −14.3 | −7.0 | −14.2 |
| Compound | S() | T() | |
|---|---|---|---|
| furan | 6.28 | 3.97/3.97/3.97 | 0.01 |
| thiophene | 5.85 | 3.74/3.74/3.74 | 0.06 |
| selenophene | 5.50 | 3.53/3.53/3.53 | 0.38 |
| CFBr⋯furan (‖) | 6.29 | 3.97/3.97/3.97 | 1.05 |
| CFBr⋯furan (⊥) | 6.35 | 4.00/4.00/4.00 | 14.32 |
| CFBr⋯furan (⊥) | 6.22 | 4.00/4.00/4.00 | 11.77 |
| CFI⋯furan (‖) | 6.31 | 4.00/4.00/4.00 | 0.09 |
| CFI⋯furan (⊥) | 6.29 | 4.03/4.03/4.04 | 23.73 |
| CFI⋯thiophene (⊥) | 5.80 | 3.80/3.80/3.80 | 41.17 |
| CFI⋯selenophene (⊥) | 5.44 | 3.61/3.61/3.61 | 68.56 |
| CFI⋯furan (⊥) | 6.24 | 4.03/4.03/4.04 | 6.84 |
| CFI⋯thiophene (⊥) | 5.85 | 3.80/3.80/3.80 | 64.62 |
| CFI⋯selenophene (⊥) | 5.49 | 3.61/3.61/3.61 | 67.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha, A.V.; Havenith, R.W.A.; van Gog, J.; De Vleeschouwer, F.; De Proft, F.; Herrebout, W. The Halogen Bond in Weakly Bonded Complexes and the Consequences for Aromaticity and Spin-Orbit Coupling. Molecules 2023, 28, 772. https://doi.org/10.3390/molecules28020772
Cunha AV, Havenith RWA, van Gog J, De Vleeschouwer F, De Proft F, Herrebout W. The Halogen Bond in Weakly Bonded Complexes and the Consequences for Aromaticity and Spin-Orbit Coupling. Molecules. 2023; 28(2):772. https://doi.org/10.3390/molecules28020772
Chicago/Turabian StyleCunha, Ana V., Remco W. A. Havenith, Jari van Gog, Freija De Vleeschouwer, Frank De Proft, and Wouter Herrebout. 2023. "The Halogen Bond in Weakly Bonded Complexes and the Consequences for Aromaticity and Spin-Orbit Coupling" Molecules 28, no. 2: 772. https://doi.org/10.3390/molecules28020772