
QuEChERS-Based Approach to the Extraction of Five Calcium Channel Blockers from Plasma Determined by UPLC-MS/MS

Abstract
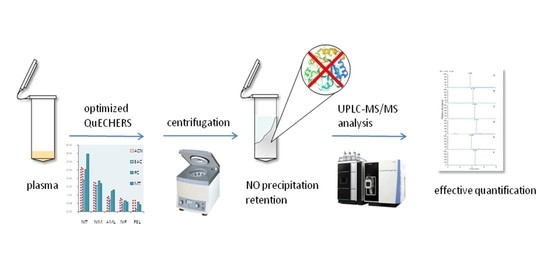
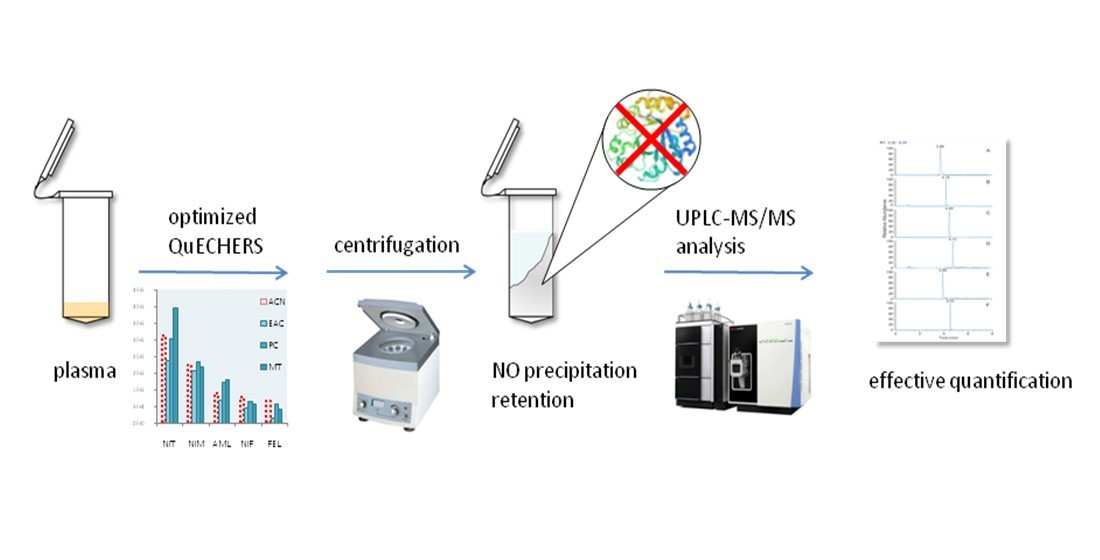
:
1. Introduction
2. Results
2.1. Methodological Validation
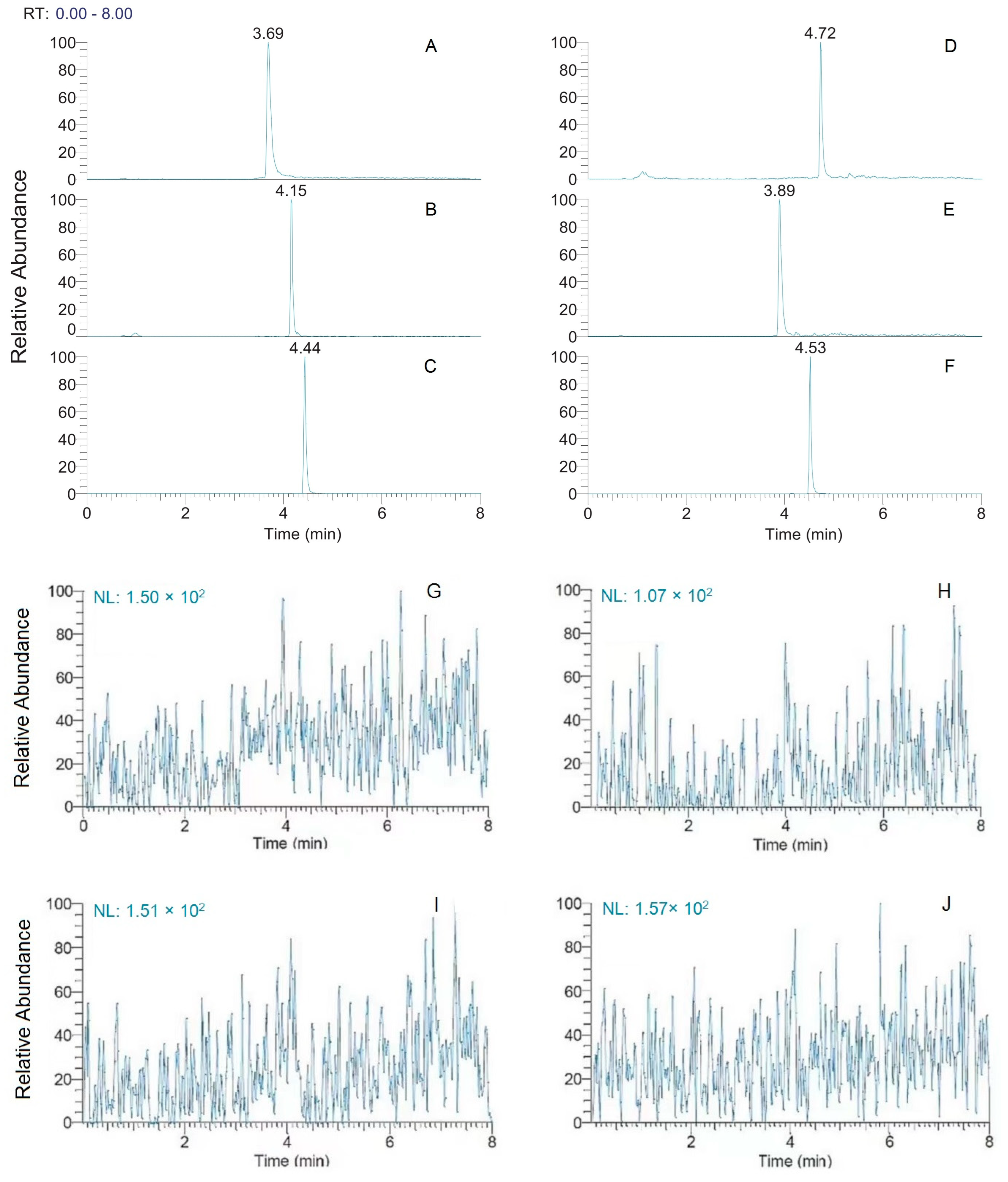
2.1.1. Selectivity
2.1.2. Linearity
2.1.3. Accuracy and Precision
2.1.4. Matrix Effect
2.1.5. Stability
3. Discussion
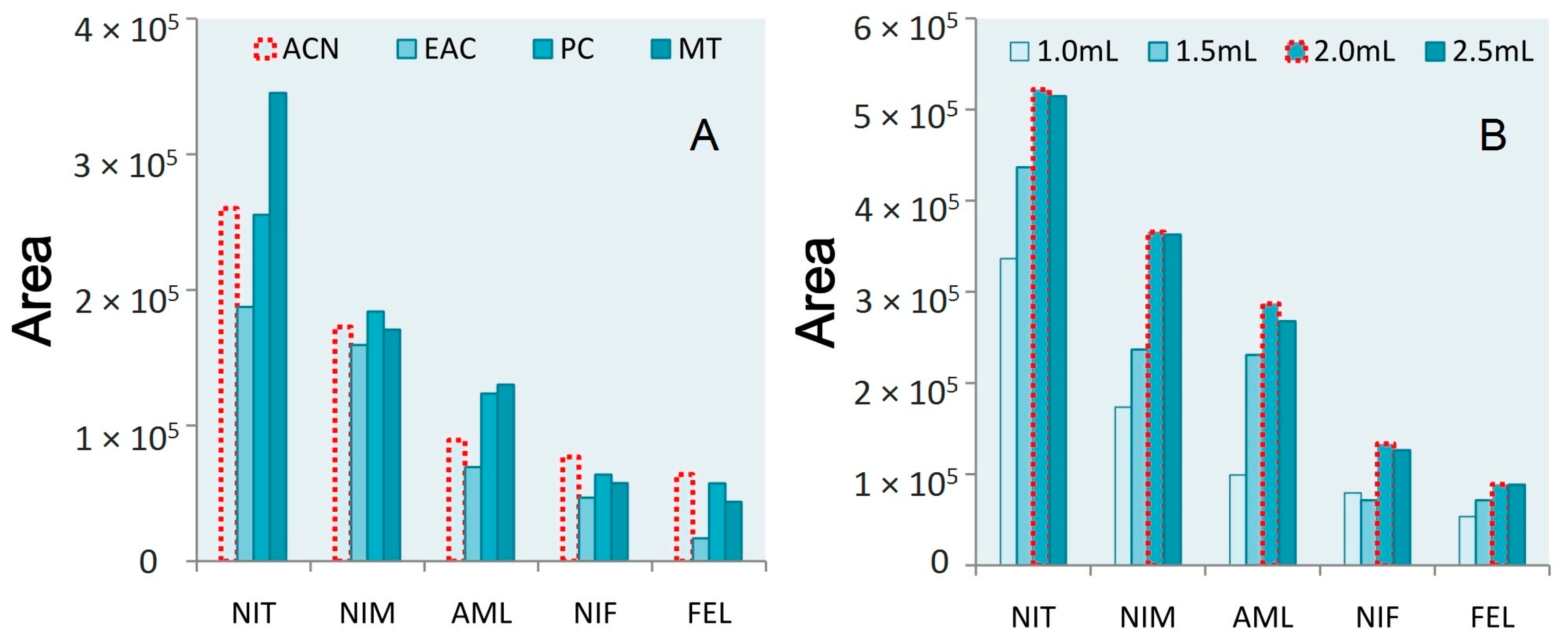
3.1. Extraction Procedure Optimization
3.2. Purification Reagent Selection
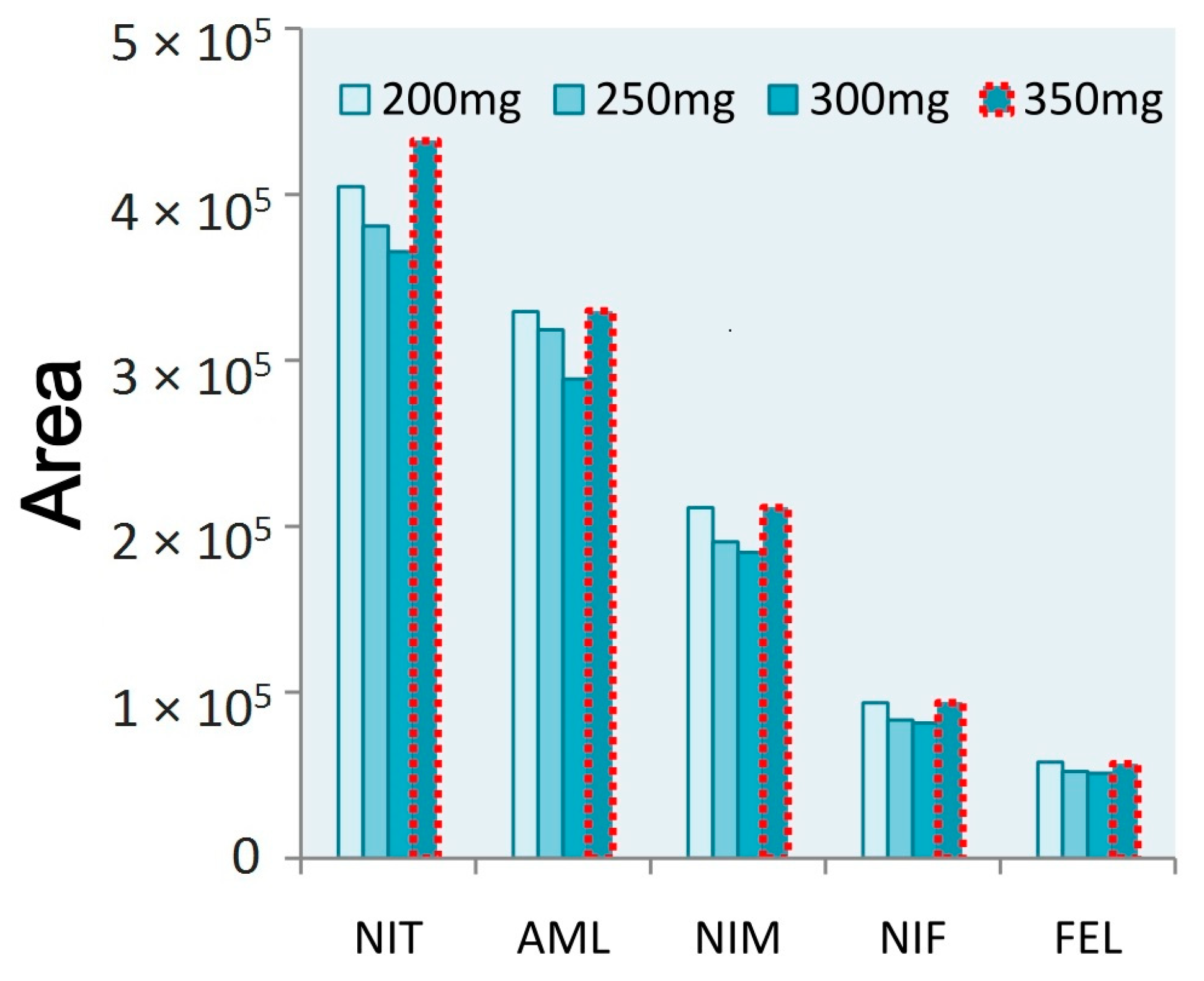
3.3. Salting Reagent Selection
3.4. Comparisons
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Instrument and Analytical Conditions
4.3. Solution Preparation
4.4. Plasma Sample Preparation
4.5. Methodological Validation
4.5.1. Selectivity and Carryover
4.5.2. Linearity
4.5.3. Precision and Accuracy
4.5.4. Stability
4.5.5. Matrix Effect and Extraction Recovery
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Gummin, D.D.; Mowry, J.B.; Spyker, D.A.; Brooks, D.E.; Beuhler, M.C.; Rivers, L.J.; Hashem, H.A.; Ryan, M.L. 2018 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 36th Annual Report. Clin. Toxicol. 2019, 57, 1220–1413. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, H.T.; Olsen, J.H.; Mellemkjaer, L.; Marie, A.; Steffensen, F.H.; McLaughlin, J.K.; Baron, J.A. Cancer risk and mortality in users of calcium channel blockers. A cohort study. Cancer 2000, 89, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Hoffman, R.S.; Walsh, S.J.; Roberts, D.M.; Gosselin, S.; Bunchman, T.E.; Kebede, S.; Lavergne, V.; Ghannoum, M. EXTRIP workgroup. Extracorporeal treatment for calcium channel blocker poisoning: Systematic review and recommendations from the EXTRIP workgroup. Clin. Toxicol. 2021, 59, 361–375. [Google Scholar] [CrossRef]
- Mladěnka, P.; Applová, L.; Patočka, J.; Costa, V.M.; Remiao, F.; Pourová, J.; Mladěnka, A.; Karlíčková, J.; Jahodář, L.; Vopršalová, M.; et al. TOX-OER and CARDIOTOX Hradec Králové Researchers and Collaborators. Comprehensive review of cardiovascular toxicity of drugs and related agents. Med. Res. Rev. 2018, 38, 1332–1403. [Google Scholar] [CrossRef] [Green Version]
- Nawrot, D.A.; Ozer, L.Y.; Al Haj Zen, A. A Novel High Content Angiogenesis Assay Reveals That Lacidipine, L-Type Calcium Channel Blocker, Induces In Vitro Vascular Lumen Expansion. Int. J. Mol. Sci. 2022, 28, 4891. [Google Scholar] [CrossRef] [PubMed]
- St-Onge, M. Cardiovascular Drug Toxicity. Crit. Care Clin. 2021, 37, 563–576. [Google Scholar] [PubMed]
- Batalis, N.I.; Harley, R.A.; Schandl, C.A. Verapamil toxicity: An unusual case report and review of the literature. Am. J. Forensic. Med. Pathol. 2007, 28, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Palatnick, W.; Jelic, T. Calcium channel blocker and beta blocker overdose, and digoxin toxicity management. Emerg. Med. Pract. 2020, 22 (Suppl. S9), 1–42. [Google Scholar]
- Levine, M.; Curry, S.C.; Padilla-Jones, A.; Ruha, A.M. Critical care management of verapamil and diltiazem overdose with a focus on vasopressors: A 25-year experience at a single center. Ann. Emerg. Med. 2013, 62, 252–258. [Google Scholar] [CrossRef]
- Chen, G.; Jirjees Sr, F.; Al Bawab, A.; McElnay, J.C. Quantification of amlodipine in dried blood spot samples by high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2018, 1072, 252–258. [Google Scholar] [CrossRef]
- Wang, W.; Li, P.; Wu, F.; Wang, Z.; Zhang, T. Development and optimization of a supercritical fluid chromatography tandem mass spectrometry method for the high-throughput determination of nimodipine in beagle plasma. J. Sep. Sci. 2019, 42, 2179–2186. [Google Scholar] [PubMed]
- Chen, M.; Zhou, J.; Mei, L.; Yu, F.; Xie, X.; Liu, Y.; Yang, Y.; Li, Y.; Mei, X. Simultaneous Determination of Felodipine and Metoprolol in Beagle Dog Plasma by Online SPE-LC-MS/MS and Its Application in a Pharmacokinetic Study. Anal. Sci. 2017, 33, 755–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammad, M.A.; Mahrouse, M.A.; Amer, E.A.H.; Elharati, N.S. Validated LC-MS/MS method for the simultaneous determination of enalapril maleate, nitrendipine, hydrochlorothiazide, and their major metabolites in human plasma. Biomed Chromatogr. 2020, 34, e4955. [Google Scholar] [CrossRef] [PubMed]
- Heidari, H.; Limouei-Khosrowshahi, B. Magnetic solid phase extraction with carbon-coated Fe3O4 nanoparticles coupled to HPLC-UV for the simultaneous determination of losartan, carvedilol, and amlodipine besylate in plasma samples. J. Chromatogr. B Analyt. Technol. Biomed Life Sci. 2019, 1114–1115, 24–30. [Google Scholar] [CrossRef]
- Jain, B.; Jain, R.; Kabir, A.; Sharma, S. Rapid Determination of Non-Steroidal Anti-Inflammatory Drugs in Urine Samples after In-Matrix Derivatization and Fabric Phase Sorptive Extraction-Gas Chromatography-Mass Spectrometry Analysis. Molecules 2022, 27, 7188. [Google Scholar] [CrossRef] [PubMed]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and “dispersive solid-phase extraction” for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Su, Y.; Liu, J.; Zhang, K.; Wang, X.; Chen, Y.; Duan, L.; Shi, F. Determination of cyflufenamid residues in 12 foodstuffs by QuEChERS-HPLC-MS/MS. Food Chem. 2021, 362, 130148. [Google Scholar] [CrossRef]
- Abdelghani, J.I.; Al-Degs, Y.S. Spectroscopic quantifiication of preservatives in different food matrices using QuEChERS extraction and multivariate calibration with comparison against liquid chromatography. Arab J. Chem. 2022, 15, 103462. [Google Scholar] [CrossRef]
- Hernández-Mesa, M.; García-Campaña, A.M. Determination of sulfonylurea pesticide residues in edible seeds used as nutraceuticals by QuEChERS in combination with ultra-high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2020, 1617, 460831. [Google Scholar] [CrossRef]
- Rejczak, T.; Tuzimski, T. A review of recent developments and trends in the QuEChERS sample preparation approach. Open Chem. 2015, 13, 980–1010. [Google Scholar] [CrossRef]
- Attala, K.; Eissa, M.S.; Hasan, M.A.; El-Henawee, M.M.; Abd El-Hay, S.S. An enhanced first derivative synchronous spectrofluorimetric method for determination of the newly co-formulated drugs, amlodipine and celecoxib in pharmaceutical preparation and human plasma. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 240, 118533. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhao, T.; Zhen, X.; Jin, C.; Li, H.; Ha, J. Rapid Determination of 9 Tyrosine Kinase Inhibitors for the Treatment of Hepatocellular Carcinoma in Human Plasma by QuEChERS-UPLC-MS/MS. Front. Pharmacol. 2022, 13, 920436. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Song, J.; Xue, H.; Li, H.; Lian, K. Simultaneous determination of six immunosuppressants in human whole blood by HPLC-MS/MS using a modified QuEChERS method. Molecules 2022, 27, 4087. [Google Scholar] [CrossRef]
- Gao, J.; Ma, C.; Duan, Q.; Gao, W.; Li, H. Simultaneous determination of 16illegally added drugs in capsule dietary supplements using a QuEChERS method and HPLC–MS/MS. Chromatographia 2021, 84, 1009–1023. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Linear Equation | Linear Range (ng·mL−1) | R2 | LODs (ng·mL−1) | LOQs (ng·mL−1) |
|---|---|---|---|---|---|
| AML | Y = −0.171406 + 0.0869037x | 2.0–80 | 0.9951 | 0.014 | 0.048 |
| FEL | Y = −0.0524462 + 0.00288808x | 20–800 | 0.9933 | 0.220 | 0.732 |
| NIF | Y = −0.429218 + 0.421058x | 1.0–40 | 0.9921 | 0.002 | 0.006 |
| NIM | Y = −0.107548 + 0.0537931x | 2.0–80 | 0.9914 | 0.007 | 0.025 |
| NIR | Y = −0.0401095 + 0.0169601x | 6.0–240 | 0.9947 | 0.041 | 0.137 |
| Compounds | Spiked (ng·mL−1) | ME (%) | ER (%) | Accuracy (%) | Precision (RSD %) | |
|---|---|---|---|---|---|---|
| Inter-Day | Intra-Day | |||||
| AML | 5 | 102.06 | 95.23 | 101.43 | 0.69 | 2.00 |
| 20 | 99.64 | 102.67 | 105.83 | 6.89 | 1.81 | |
| 80 | 93.59 | 94.06 | 87.54 | 2.24 | 2.86 | |
| FEL | 50 | 97.18 | 106.17 | 103.95 | 3.35 | 0.64 |
| 200 | 94.52 | 94.86 | 106.15 | 11.04 | 2.87 | |
| 800 | 91.21 | 96.41 | 90.15 | 6.36 | 2.61 | |
| NIF | 2.5 | 91.23 | 100.80 | 111.23 | 0.82 | 0.28 |
| 10 | 98.19 | 95.98 | 96.27 | 3.79 | 0.71 | |
| 40 | 92.43 | 91.74 | 91.60 | 4.45 | 2.10 | |
| NIM | 5 | 93.66 | 91.94 | 104.18 | 0.21 | 1.45 |
| 20 | 110.48 | 95.18 | 103.35 | 8.51 | 1.85 | |
| 80 | 95.09 | 97.45 | 92.91 | 5.71 | 11.64 | |
| NIT | 15 | 99.11 | 97.55 | 113.05 | 4.43 | 2.57 |
| 60 | 116.39 | 98.45 | 105.08 | 0.19 | 1.66 | |
| 240 | 108.84 | 94.78 | 96.63 | 3.22 | 2.75 | |
| Compounds | Spiked (ng·mL−1) | Stability (RSD%) | |||
|---|---|---|---|---|---|
| 25 °C/24 h | 4 °C/24 h | 4 °C/48 h | −20 °C/7d | ||
| AML | 5 | 1.52 | 1.44 | 2.69 | 6.33 |
| 20 | 2.30 | 1.61 | 0.95 | 0.69 | |
| 80 | 1.50 | 0.48 | 1.62 | 3.36 | |
| FEL | 50 | 6.11 | 2.13 | 4.77 | 5.38 |
| 200 | 1.96 | 3.19 | 7.00 | 4.72 | |
| 800 | 5.47 | 0.85 | 2.45 | 9.41 | |
| NIF | 2.5 | 2.16 | 1.43 | 1.64 | 2.34 |
| 10 | 7.42 | 0.57 | 0.92 | 1.23 | |
| 40 | 0.91 | 1.26 | 0.37 | 4.53 | |
| NIM | 5 | 4.95 | 1.70 | 5.58 | 2.93 |
| 20 | 3.02 | 1.20 | 2.40 | 4.50 | |
| 80 | 0.50 | 5.41 | 3.27 | 7.60 | |
| NIT | 15 | 2.16 | 1.70 | 1.55 | 8.08 |
| 60 | 10.05 | 1.06 | 6.42 | 2.02 | |
| 240 | 2.50 | 3.64 | 2.75 | 9.10 | |
| Method | Analyte | LOD(ng/mL) | LOQ(ng/mL) | ER(%) | Ref. |
|---|---|---|---|---|---|
| QuEChERS/UPLC-MS/MS | AML | 0.014 | 0.04 | 97.32 | This work |
| FEL | 0.220 | 80.732 | 96.15 | ||
| NIF | 0.002 | 0.006 | 96.17 | ||
| NIM | 0.007 | 0.025 | 94.85 | ||
| NIT | 0.041 | 0.137 | 96.93 | ||
| PP/first derivative SFS | AML | 1.160 | 3.516 | 95.20 | [21] |
| HPLC-MS/MS | AML | 0.2 | 0.5 | 50.90 | [11] |
| MSPE/HPLC-UV | NIM | 0.28 | 0.84 | 59.87 | [12] |
| PP/SFC-MS/MS | NIM | 0.05 | 0.12 | 91 | [13] |
| SPE/LC-MS/MS | FEL | - | 0.59 | 91.9 | [14] |
| LLE/LC-MS/MS | NIT | - | - | 89.51 | [15] |
| Compounds | Precursor (m/z) | Product (m/z) | Collision Energy(V) | RFLens(V) | Retention Time (min) |
|---|---|---|---|---|---|
| AML | 409.200 | 237.9 | 12.03 | 53 | 3.89 |
| 294.0 | 12.20 | ||||
| FEL | 385.175 | 339.0 | 13.13 | 53 | 4.72 |
| 353.0 | 13.55 | ||||
| NIF | 347.175 | 264.9 | 12.75 | 46 | 4.15 |
| 314.9 | 8.79 | ||||
| NIM | 419.212 | 301.1 | 20.60 | 55 | 4.53 |
| 343.1 | 10.64 | ||||
| NIR | 361.262 | 315.1 | 13.17 | 64 | 4.44 |
| 329.1 | 13.72 | ||||
| PRO | 260.175 | 155.0 | 25.30 | 47 | 3.69 |
| 157.0 | 20.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, T.; Jiang, W.; Zhen, X.; Jin, C.; Zhang, Y.; Li, H. QuEChERS-Based Approach to the Extraction of Five Calcium Channel Blockers from Plasma Determined by UPLC-MS/MS. Molecules 2023, 28, 671. https://doi.org/10.3390/molecules28020671
Zhao T, Jiang W, Zhen X, Jin C, Zhang Y, Li H. QuEChERS-Based Approach to the Extraction of Five Calcium Channel Blockers from Plasma Determined by UPLC-MS/MS. Molecules. 2023; 28(2):671. https://doi.org/10.3390/molecules28020671
Chicago/Turabian StyleZhao, Tingting, Wen Jiang, Xiaolan Zhen, Chengcheng Jin, Yifan Zhang, and Hui Li. 2023. "QuEChERS-Based Approach to the Extraction of Five Calcium Channel Blockers from Plasma Determined by UPLC-MS/MS" Molecules 28, no. 2: 671. https://doi.org/10.3390/molecules28020671