
Interlaboratory Evaluation of a User-Friendly Benchtop Mass Spectrometer for Multiple-Attribute Monitoring Studies of a Monoclonal Antibody

 , , , and
, , , and 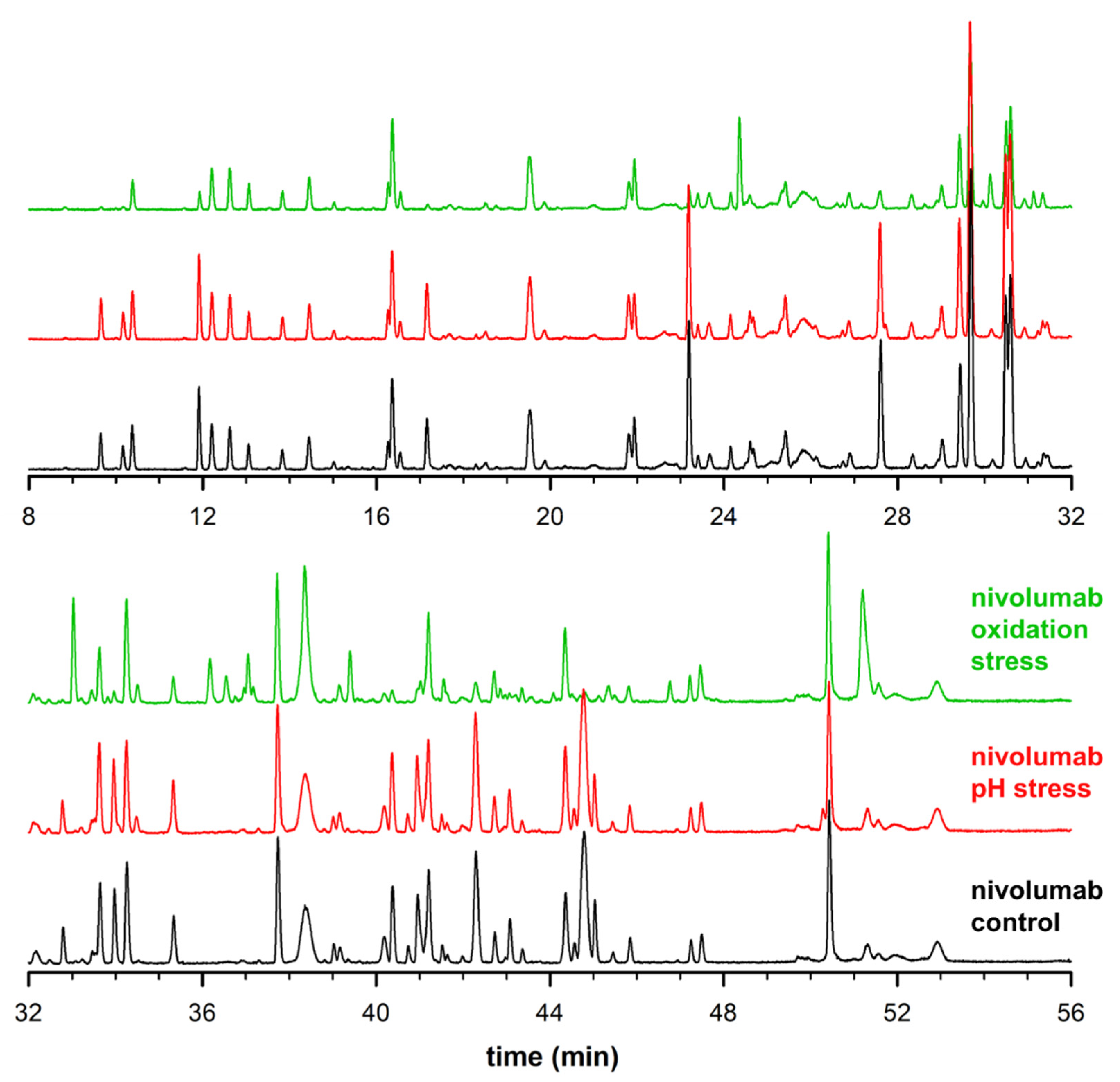{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Study Design
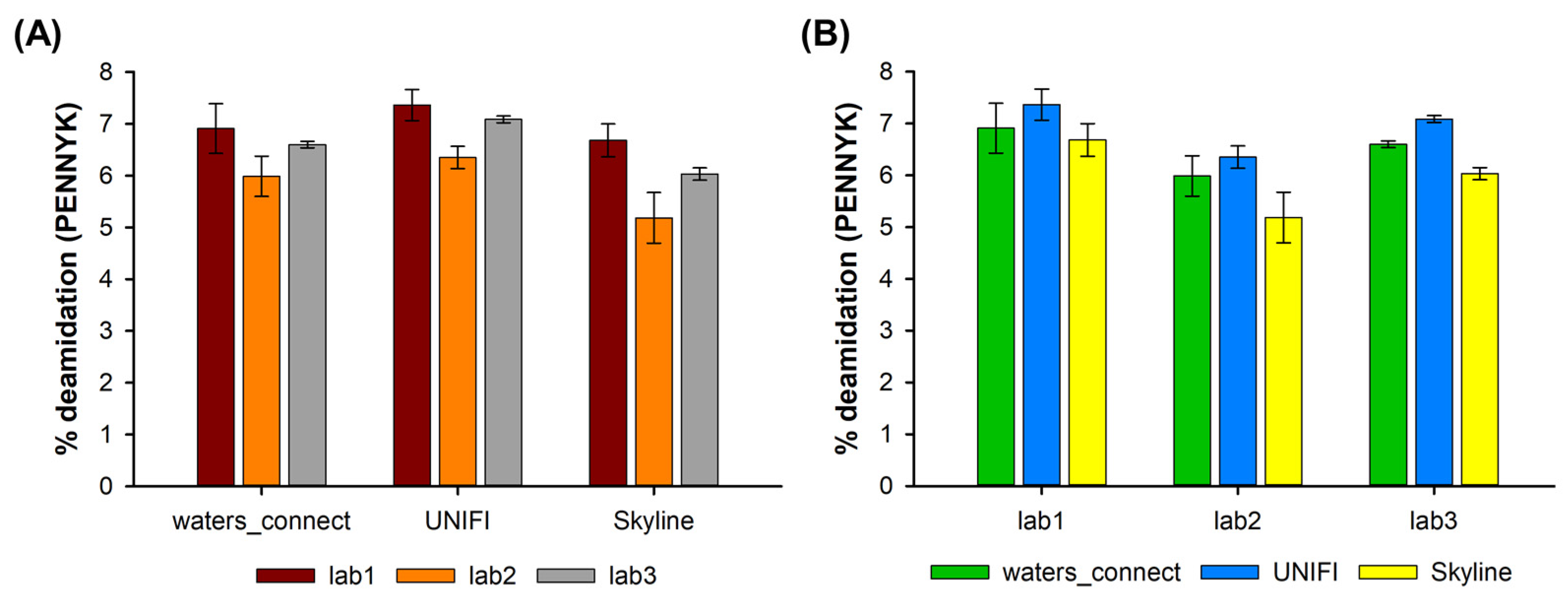
2.2. Asparagine Deamidation
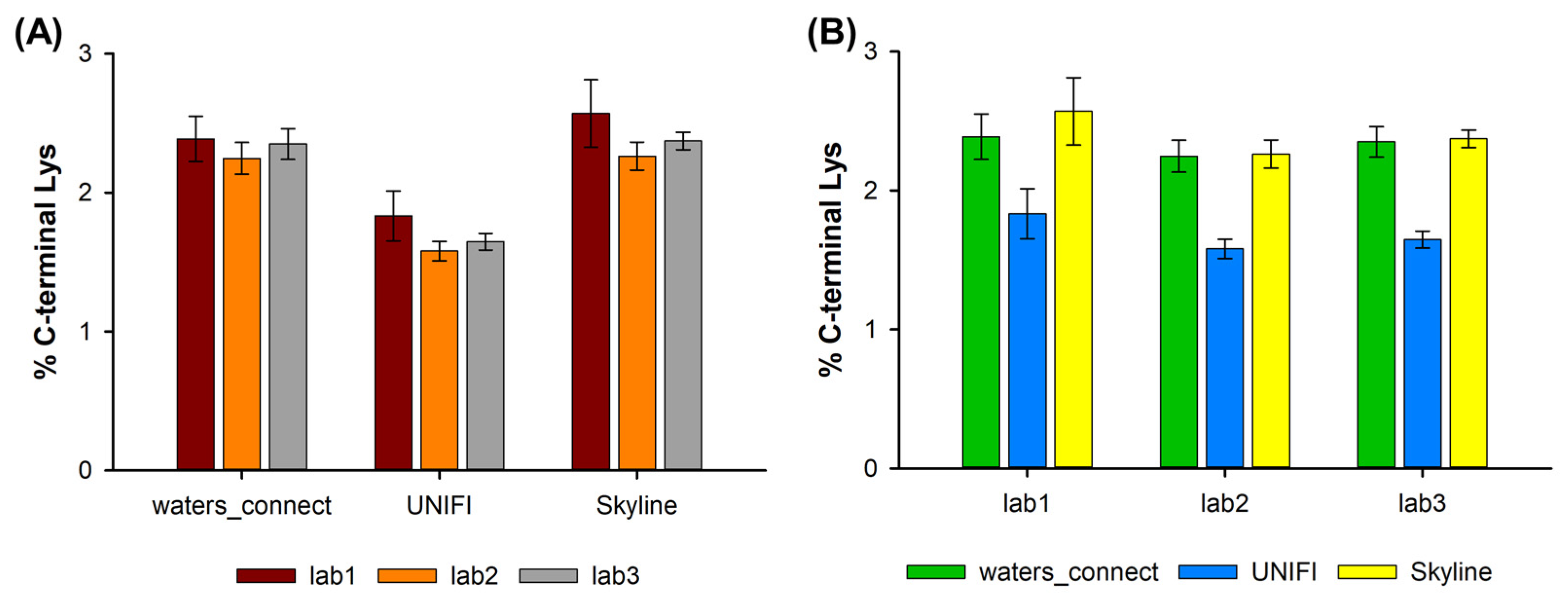
2.3. Presence of C-Terminal Lysine
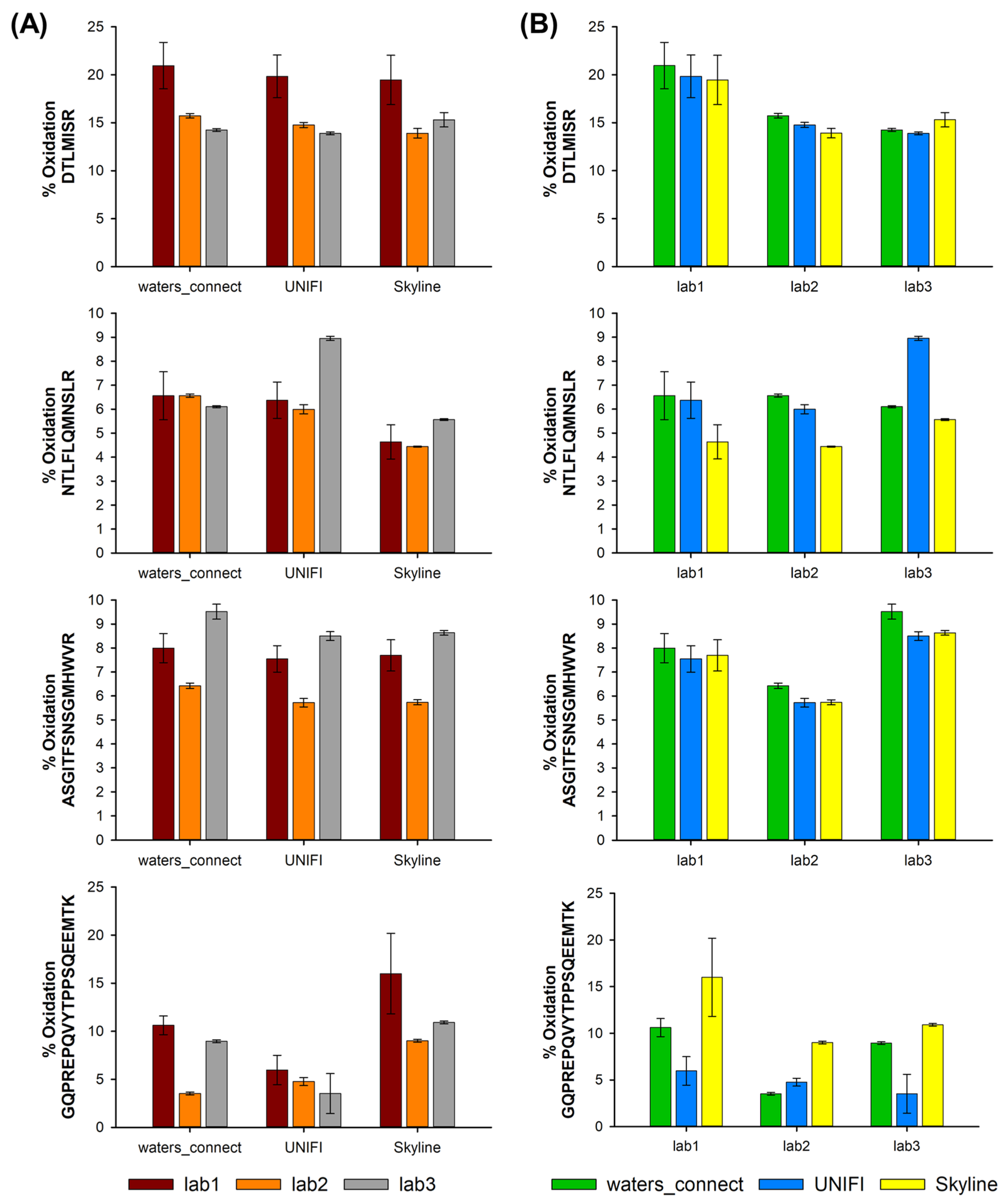
2.4. Methionine Oxidation
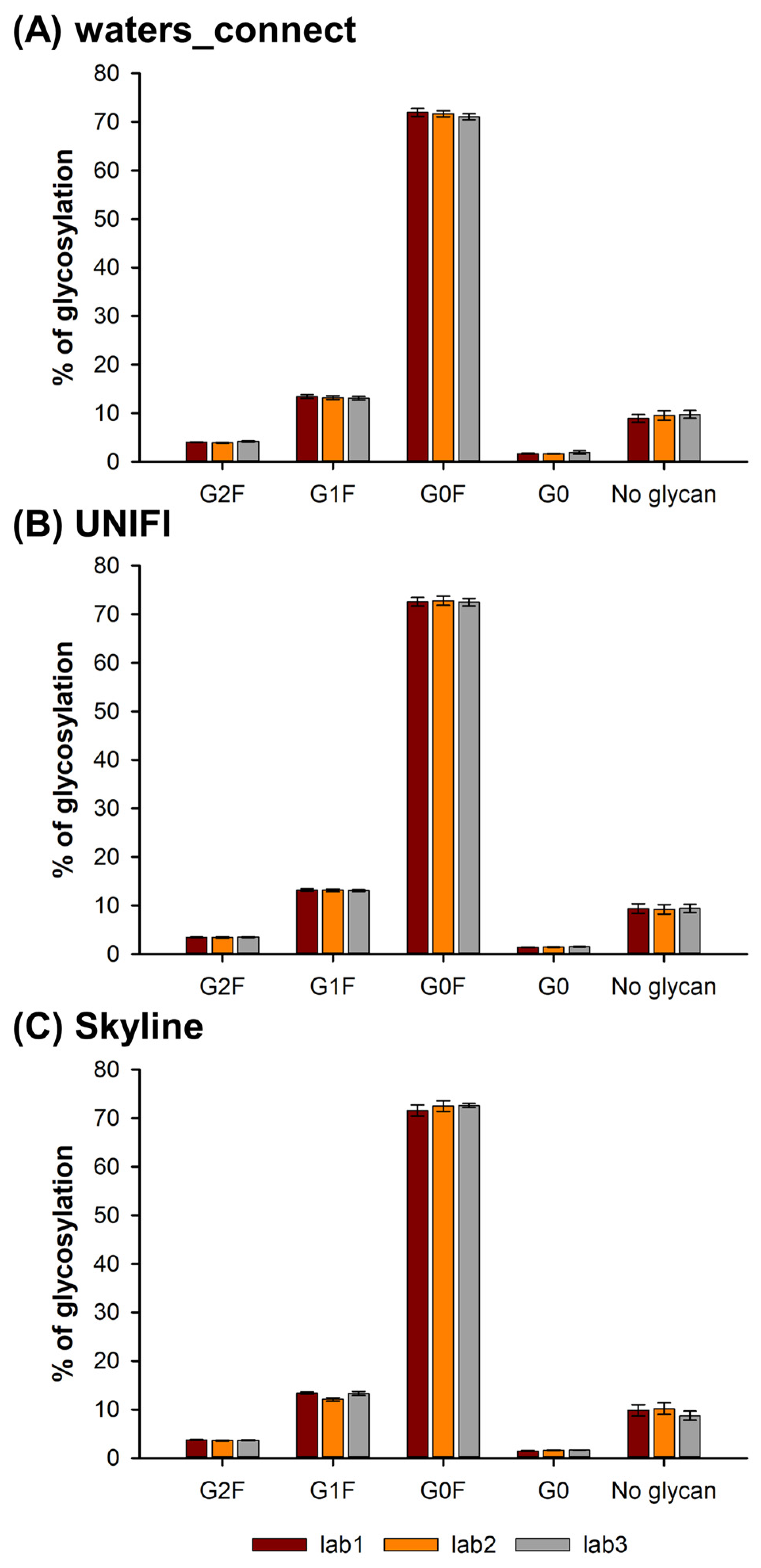
2.5. Glycosylation
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Sample Preparation
3.3. Generic Chromatographic Conditions
3.4. LC–MS Instrumentation and MS Settings
3.5. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Mouchahoir, T.; Schiel, J.E. Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Anal. Bioanal. Chem. 2018, 410, 2111–2126. [Google Scholar] [CrossRef] [Green Version]
- Michels, D.A.; Ip, A.Y.; Dillon, T.M.; Brorson, K.; Lute, S.; Chavez, B.; Prentice, K.M.; Brady, L.J.; Miller, K.J. Separation Methods and Orthogonal Techniques. In State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2—Biopharmaceutical Characterization: The NISTmAb Case Study; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2015; pp. 237–284. [Google Scholar]
- Li, W.; Kerwin, J.L.; Schiel, J.; Formolo, T.; Davis, D.; Mahan, A.; Benchaar, S.A. Structural Elucidation of Post-Translational Modifications in Monoclonal Antibodies. In State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2—Biopharmaceutical Characterization: The NISTmAb Case Study; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2015; Volume 1201, pp. 119–183. [Google Scholar]
- Hao, Z.; Moore, B.; Ren, C.; Sadek, M.; Macchi, F.; Yang, L.; Harris, J.; Yee, L.; Liu, E.; Tran, V.; et al. Multi-attribute method performance profile for quality control of monoclonal antibody therapeutics. J. Pharm. Biomed. Anal. 2021, 205, 114330. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.S.; Nightlinger, N.S.; Livingston, B.; Campbell, P.; Bailey, R.; Balland, A. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. mAbs 2015, 7, 881–890. [Google Scholar] [CrossRef]
- Mouchahoir, T.; Schiel, J.E.; Rogers, R.; Heckert, A.; Place, B.J.; Ammerman, A.; Li, X.; Robinson, T.; Schmidt, B.; Chumsae, C.M.; et al. New Peak Detection Performance Metrics from the MAM Consortium Interlaboratory Study. J. Am. Soc. Mass Spectrom. 2021, 32, 913–928. [Google Scholar] [CrossRef]
- Khawli, L.A.; Goswami, S.; Hutchinson, R.; Kwong, Z.W.; Yang, J.; Wang, X.; Yao, Z.; Sreedhara, A.; Cano, T.; Tesar, D.; et al. Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats. mAbs 2010, 2, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Chu, L.; Li, W.; Lawson, K.; Apostol, I.; Eris, T. Application of a Quantitative LC–MS Multiattribute Method for Monitoring Site-Specific Glycan Heterogeneity on a Monoclonal Antibody Containing Two N-Linked Glycosylation Sites. Anal. Chem. 2017, 89, 3562–3567. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Qiu, H.; Li, N. LC-MS multi-attribute method for characterization of biologics. J. Appl. Bioanal. 2017, 3, 21–25. [Google Scholar] [CrossRef]
- Carvalho, S.B.; Gomes, R.A.; Pfenninger, A.; Fischer, M.; Strotbek, M.; Isidro, I.A.; Tugcu, N.; Gomes-Alves, P. Multi attribute method implementation using a High Resolution Mass Spectrometry platform: From sample preparation to batch analysis. PLoS ONE 2022, 17, e0262711. [Google Scholar] [CrossRef] [PubMed]
- Sitasuwan, P.; Powers, T.W.; Medwid, T.; Huang, Y.; Bare, B.; Lee, L.A. Enhancing the multi-attribute method through an automated and high-throughput sample preparation. mAbs 2021, 13, 1978131. [Google Scholar] [CrossRef] [PubMed]
- Rogstad, S.; Yan, H.; Wang, X.; Powers, D.; Brorson, K.; Damdinsuren, B.; Lee, S. Multi-Attribute Method for Quality Control of Therapeutic Proteins. Anal. Chem. 2019, 91, 14170–14177. [Google Scholar] [CrossRef]
- Ogata, Y.; Quizon, P.M.; Nightlinger, N.S.; Sitasuwan, P.; Snodgrass, C.; Lee, L.A.; Meyer, J.D.; Rogers, R.S. Automated multi-attribute method sample preparation using high-throughput buffer exchange tips. Rapid Commun. Mass Spectrom. 2022, 36, e9222. [Google Scholar] [CrossRef] [PubMed]
- Rathore, D.; Faustino, A.; Schiel, J.; Pang, E.; Boyne, M.; Rogstad, S. The role of mass spectrometry in the characterization of biologic protein products. Expert. Rev. Proteom. 2018, 15, 431–449. [Google Scholar] [CrossRef] [PubMed]
- Ren, D. Advancing Mass Spectrometry Technology in cGMP Environments. Trends Biotechnol. 2020, 38, 1051–1053. [Google Scholar] [CrossRef]
- Zhang, A.; Chen, Z.; Li, M.; Qiu, H.; Lawrence, S.; Bak, H.; Li, N. A general evidence-based sequence variant control limit for recombinant therapeutic protein development. mAbs 2020, 12, 1791399. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.B.; Orgaard, M.; Sloth, T.M.; Christoffersen, N.S.; Leth-Espensen, K.Z.; Jensen, P.F. Optimized Multi-Attribute Method Workflow Addressing Missed Cleavages and Chromatographic Tailing/Carry-Over of Hydrophobic Peptides. Anal. Chem. 2022, 94, 17195–17204. [Google Scholar] [CrossRef]
- Lyu, X.; Zhao, Q.; Hui, J.; Wang, T.; Lin, M.; Wang, K.; Zhang, J.; Shentu, J.; Dalby, P.A.; Zhang, H.; et al. The global landscape of approved antibody therapies. Antib. Ther. 2022, 5, 233–257. [Google Scholar] [CrossRef]
- Hernandez-Alba, O.; Wagner-Rousset, E.; Beck, A.; Cianferani, S. Native Mass Spectrometry, Ion Mobility, and Collision-Induced Unfolding for Conformational Characterization of IgG4 Monoclonal Antibodies. Anal. Chem. 2018, 90, 8865–8872. [Google Scholar] [CrossRef]
- Suckau, D.; Evers, W.; Belau, E.; Pengelley, S.; Resemann, A.; Tang, W.; Sen, K.I.; Wagner, E.; Colas, O.; Beck, A. Use of PASEF for Accelerated Protein Sequence Confirmation and De Novo Sequencing with High Data Quality. In Therapeutic Antibodies: Methods and Protocols; Houen, G., Ed.; Springer: New York, NY, USA, 2022; pp. 207–217. [Google Scholar]
- Beck, A.; Wagner-Rousset, E.; Ayoub, D.; Van Dorsselaer, A.; Sanglier-Cianferani, S. Characterization of therapeutic antibodies and related products. Anal. Chem. 2013, 85, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Largy, E.; Cantais, F.; Van Vyncht, G.; Beck, A.; Delobel, A. Orthogonal liquid chromatography–mass spectrometry methods for the comprehensive characterization of therapeutic glycoproteins, from released glycans to intact protein level. J. Chromatogr. A 2017, 1498, 128–146. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutierrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Alosert, H.; Savery, J.; Rheaume, J.; Cheeks, M.; Turner, R.; Spencer, C.; Suzane, S.F.; Goldrick, S. Data integrity within the biopharmaceutical sector in the era of Industry 4.0. Biotechnol. J. 2022, 17, e2100609. [Google Scholar] [CrossRef] [PubMed]
- Charoo, N.A.; Khan, M.A.; Rahman, Z. Data integrity issues in pharmaceutical industry: Common observations, challenges and mitigations strategies. Int. J. Pharm. 2023, 631, 122503. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.J.; Buchanan, A.; Andrews, J.; Chodorge, M.; Sridharan, S.; Mitchell, L.; Burmeister, N.; Kippen, A.D.; Vaughan, T.J.; Higazi, D.R.; et al. Rate of Asparagine Deamidation in a Monoclonal Antibody Correlating with Hydrogen Exchange Rate at Adjacent Downstream Residues. Anal. Chem. 2017, 89, 2361–2368. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Jiskoot, W.; Schoneich, C.; Rathore, A.S. Oxidation and Deamidation of Monoclonal Antibody Products: Potential Impact on Stability, Biological Activity, and Efficacy. J. Pharm. Sci. 2022, 111, 903–918. [Google Scholar] [CrossRef]
- Nowak, C.; Cheung, J.K.; Dellatore, S.M.; Katiyar, A.; Bhat, R.; Sun, J.; Ponniah, G.; Neill, A.; Mason, B.; Beck, A.; et al. Forced degradation of recombinant monoclonal antibodies: A practical guide. mAbs 2017, 9, 1217–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dick, L.W., Jr.; Qiu, D.; Mahon, D.; Adamo, M.; Cheng, K.C. C-terminal lysine variants in fully human monoclonal antibodies: Investigation of test methods and possible causes. Biotechnol. Bioeng. 2008, 100, 1132–1143. [Google Scholar] [CrossRef]
- Delobel, A. Glycosylation of Therapeutic Proteins: A Critical Quality Attribute. In Mass Spectrometry of Glycoproteins: Methods and Protocols; Methods in Molecular Biology; Delobel, A., Ed.; Springer: Clifton, NJ, USA, 2021; Volume 2271, pp. 1–21. [Google Scholar]
- Duivelshof, B.L.; Jiskoot, W.; Beck, A.; Veuthey, J.-L.; Guillarme, D.; D’Atri, V. Glycosylation of biosimilars: Recent advances in analytical characterization and clinical implications. Anal. Chim. Acta 2019, 1089, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Luo, S.; Zhang, B. Glycan analysis of therapeutic glycoproteins. mAbs 2016, 8, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Giorgetti, J.; D’Atri, V.; Canonge, J.; Lechner, A.; Guillarme, D.; Colas, O.; Wagner-Rousset, E.; Beck, A.; Leize-Wagner, E.; François, Y.-N. Monoclonal antibody N-glycosylation profiling using capillary electrophoresis—Mass spectrometry: Assessment and method validation. Talanta 2018, 178, 530–537. [Google Scholar] [CrossRef] [Green Version]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef] [Green Version]
- Schiel, J.E.; Davis, D.L.; Borisov, O.V. (Eds.) State-of-the-Art and Emerging Technologies for Therapeutic Monoclonal Antibody Characterization Volume 2. Biopharmaceutical Characterization: The NISTmAb Case Study; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2015. [Google Scholar]
- Puranik, A.; Rasam, P.; Dandekar, P.; Jain, R. Development and optimization of a LC-MS based multi-attribute method (MAM) workflow for characterization of therapeutic Fc-fusion protein. Anal. Biochem. 2023, 660, 114969. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.R.; Hebert, A.S.; Mulholland, J.; Lewis, M.J.; Hu, P. ID-MAM: A validated identity and multi-attribute monitoring method for commercial release and stability testing of a bispecific antibody. Anal. Chem. 2021, 93, 9166–9173. [Google Scholar] [CrossRef] [PubMed]
- Carillo, S.; Criscuolo, A.; Fussl, F.; Cook, K.; Bones, J. Intact multi-attribute method (iMAM): A flexible tool for the analysis of monoclonal antibodies. Eur. J. Pharm. Biopharm. 2022, 177, 241–248. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butré, C.I.; D’Atri, V.; Diemer, H.; Colas, O.; Wagner, E.; Beck, A.; Cianferani, S.; Guillarme, D.; Delobel, A. Interlaboratory Evaluation of a User-Friendly Benchtop Mass Spectrometer for Multiple-Attribute Monitoring Studies of a Monoclonal Antibody. Molecules 2023, 28, 2855. https://doi.org/10.3390/molecules28062855
Butré CI, D’Atri V, Diemer H, Colas O, Wagner E, Beck A, Cianferani S, Guillarme D, Delobel A. Interlaboratory Evaluation of a User-Friendly Benchtop Mass Spectrometer for Multiple-Attribute Monitoring Studies of a Monoclonal Antibody. Molecules. 2023; 28(6):2855. https://doi.org/10.3390/molecules28062855
Chicago/Turabian StyleButré, Claire I., Valentina D’Atri, Hélène Diemer, Olivier Colas, Elsa Wagner, Alain Beck, Sarah Cianferani, Davy Guillarme, and Arnaud Delobel. 2023. "Interlaboratory Evaluation of a User-Friendly Benchtop Mass Spectrometer for Multiple-Attribute Monitoring Studies of a Monoclonal Antibody" Molecules 28, no. 6: 2855. https://doi.org/10.3390/molecules28062855