

A Tailored Strategy to Crosslink the Aspartate Transcarbamoylase Domain of the Multienzymatic Protein CAD

Abstract
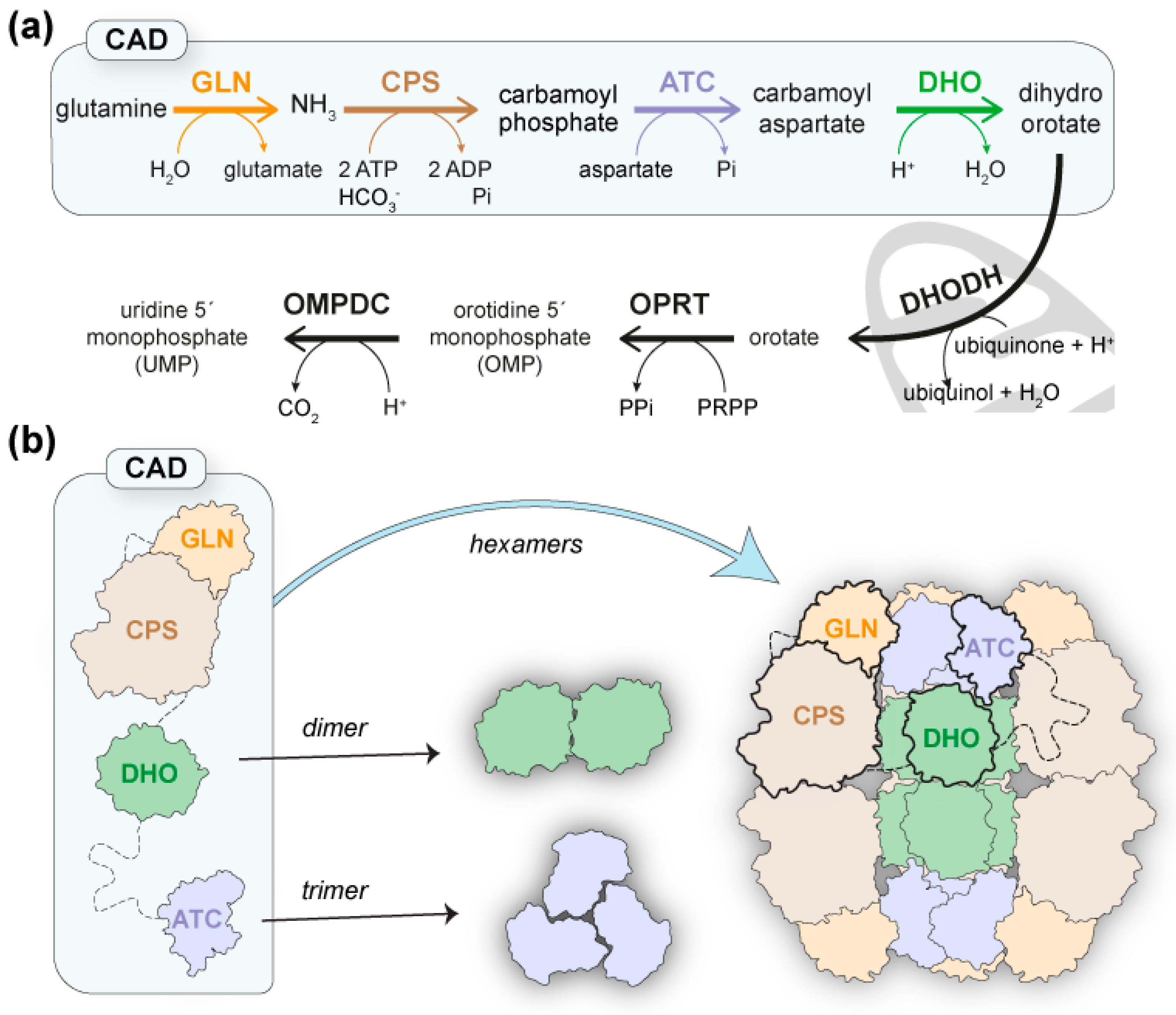
:1. Introduction
2. Results
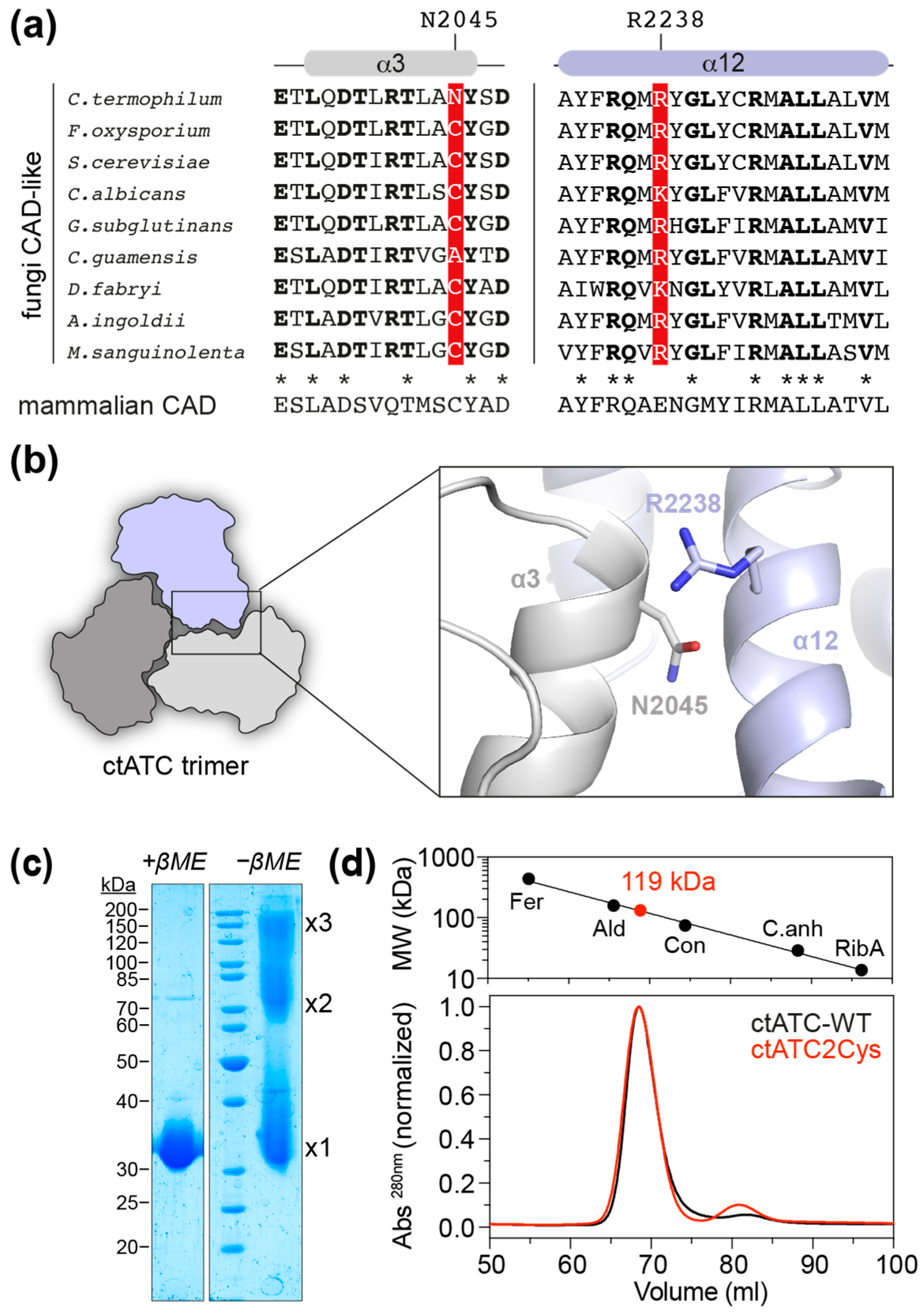
2.1. Mutant Production
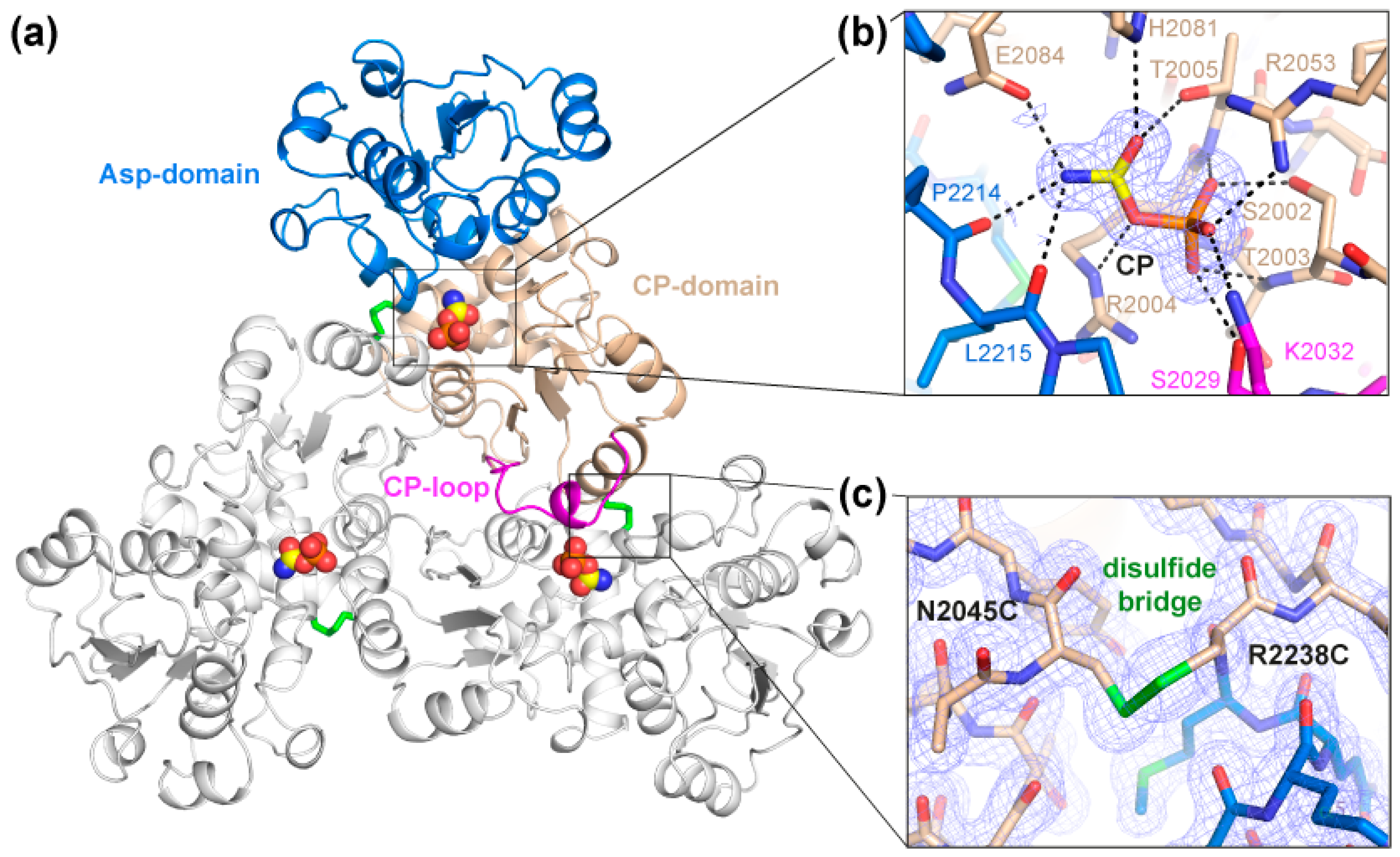
2.2. Crystal Structure of the Crosslinked ctATC2Cys Trimer
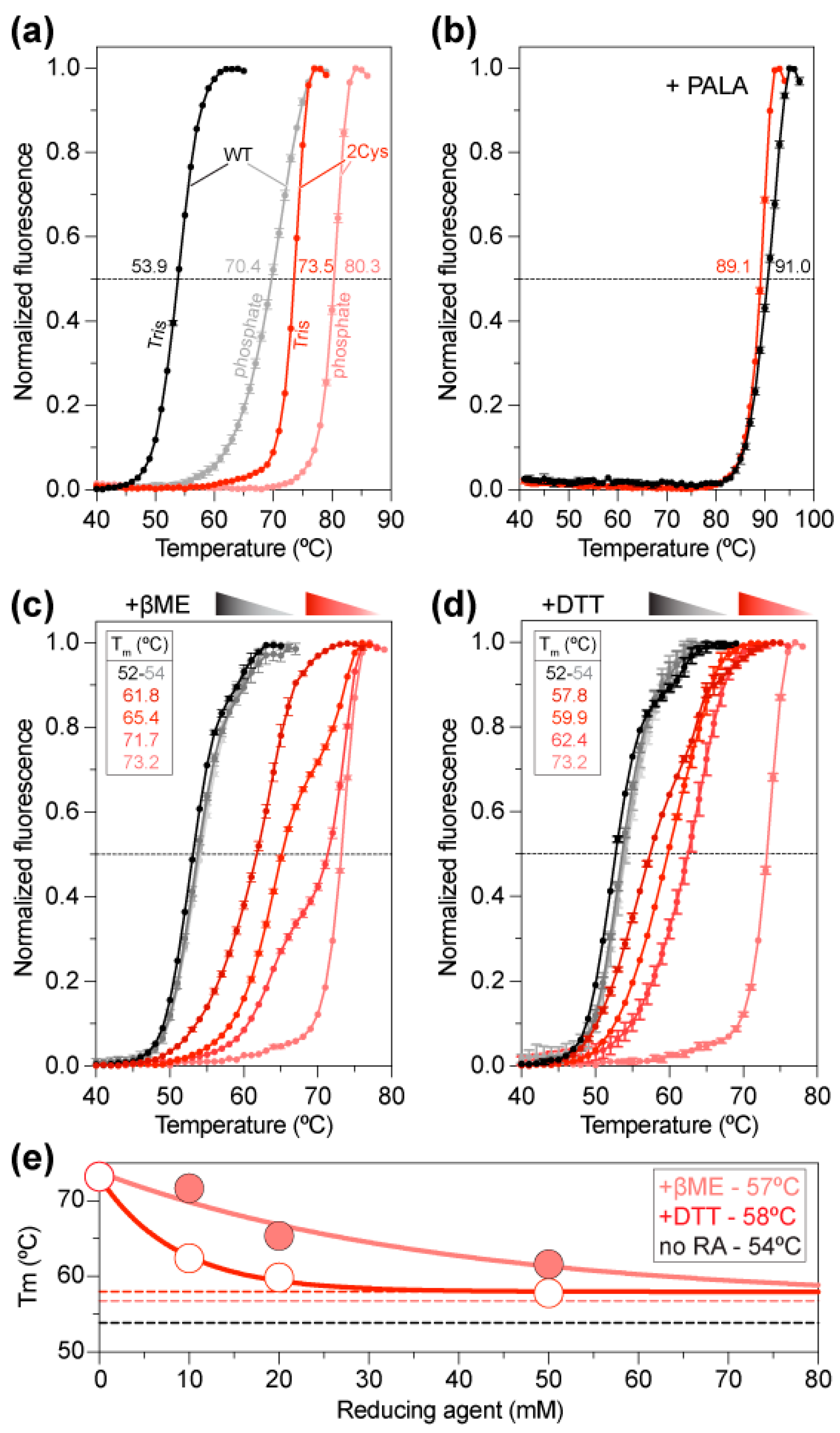
2.3. Increased Stability of the ctATC2Cys Trimer
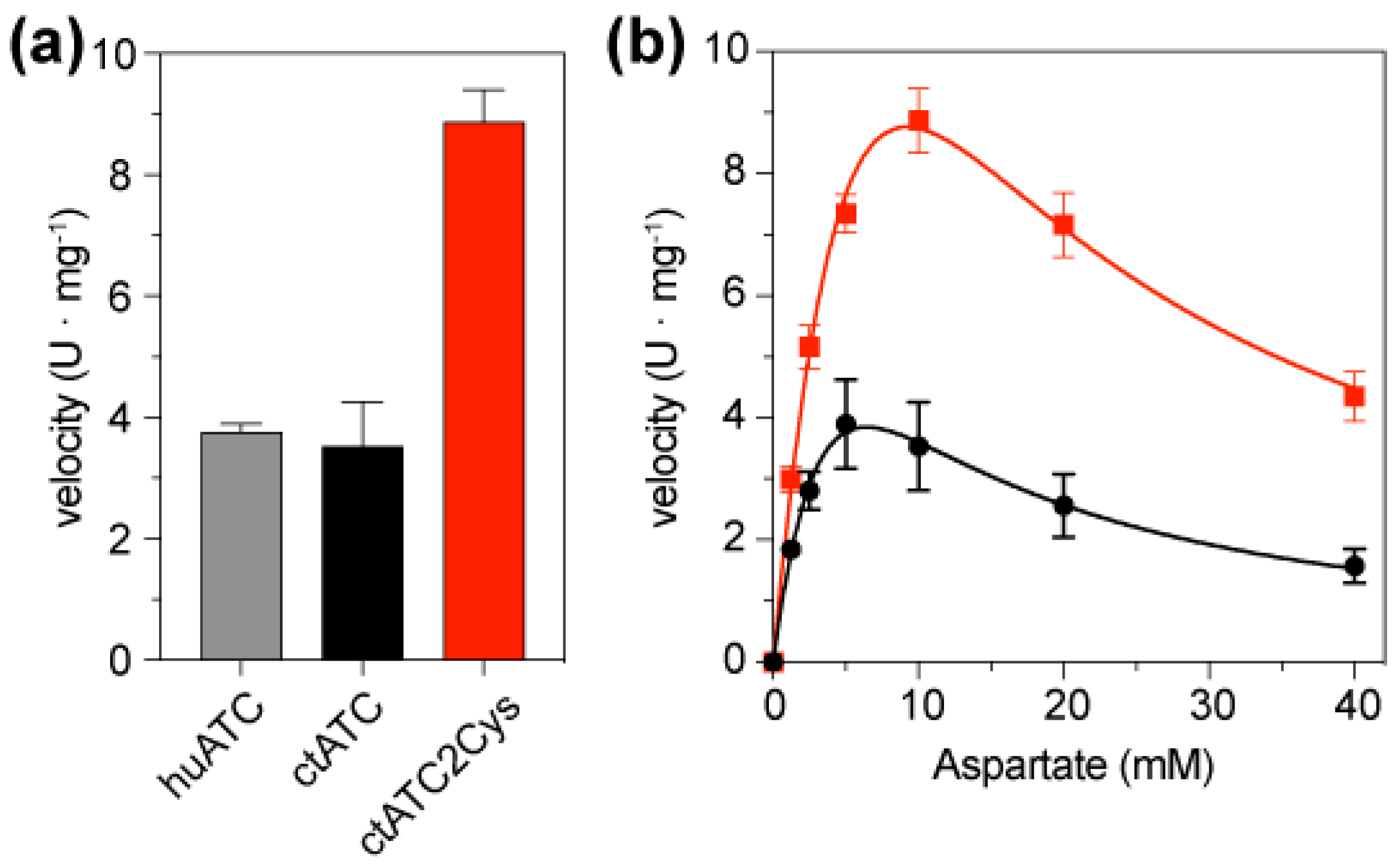
2.4. Cysteine Bridges Do Not Hinder Enzymatic Activity
3. Discussion
4. Materials and Methods
4.1. Cloning and Site-Directed Mutagenesis
4.2. Protein Expression
4.3. Protein Purification
4.4. Crystallization
4.5. Data Collection and Structure Determination
4.6. Differential Scanning Fluorimetry (DSF)
4.7. Activity Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Jones, M.E. Pyrimidine Nucleotide Biosynthesis in Animals: Genes, Enzymes, and Regulation of UMP Biosynthesis. Annu. Rev. Biochem. 1980, 49, 253–279. [Google Scholar] [CrossRef] [PubMed]
- Del Caño-Ochoa, F.; Ramón-Maiques, S. Deciphering CAD: Structure and Function of a Mega-Enzymatic Pyrimidine Factory in Health and Disease. Protein Sci. 2021, 30, 1995–2008. [Google Scholar] [CrossRef]
- Coleman, P.F.; Suttle, D.P.; Stark, G.R. Purification from Hamster Cells of the Multifunctional Protein That Initiates de Novo Synthesis of Pyrimidine Nucleotides. J. Biol. Chem. 1977, 252, 6379–6385. [Google Scholar] [CrossRef] [PubMed]
- Souciet, J.L.; Nagy, M.; Le Gouar, M.; Lacroute, F.; Potier, S. Organization of the Yeast URA2 Gene: Identification of a Defective Dihydroorotase-like Domain in the Multifunctional Carbamoylphosphate Synthetase-Aspartate Transcarbamylase Complex. Gene 1989, 79, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Denis-Duphil, M. Pyrimidine Biosynthesis in Saccharomyces Cerevisiae: The Ura2 Cluster Gene, Its Multifunctional Enzyme Product, and Other Structural or Regulatory Genes Involved in de Novo UMP Synthesis. Biochem. Cell Biol. 1989, 67, 612–631. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Guy, H.I.; Evans, D.R. Identification of the Regulatory Domain of the Mammalian Multifunctional Protein CAD by the Construction of an Escherichia Coli Hamster Hybrid Carbamyl-Phosphate Synthetase. J. Biol. Chem. 1994, 269, 27747–27755. [Google Scholar] [CrossRef]
- Carrey, E.A. Key Enzymes in the Biosynthesis of Purines and Pyrimidines: Their Regulation by Allosteric Effectors and by Phosphorylation. Biochem. Soc. Trans. 1995, 23, 899–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graves, L.M.; Guy, H.I.; Kozlowski, P.; Huang, M.; Lazarowski, E.; Pope, R.M.; Collins, M.A.; Dahlstrand, E.N.; Earp, H.S.; Evans, D.R. Regulation of Carbamoyl Phosphate Synthetase by MAP Kinase. Nature 2000, 403, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Sigoillot, F.D.; Berkowski, J.A.; Sigoillot, S.M.; Kotsis, D.H.; Guy, H.I. Cell Cycle-Dependent Regulation of Pyrimidine Biosynthesis. J. Biol. Chem. 2003, 278, 3403–3409. [Google Scholar] [CrossRef] [Green Version]
- Aoki, T.; Weber, G. Carbamoyl Phosphate Synthetase (Glutamine-Hydrolyzing): Increased Activity in Cancer Cells. Science 1981, 212, 463–465. [Google Scholar] [CrossRef]
- Swyryd, E.A.; Seaver, S.S.; Stark, G.R. N-(Phosphonacetyl)-L-Aspartate, a Potent Transition State Analog Inhibitor of Aspartate Transcarbamylase, Blocks Proliferation of Mammalian Cells in Culture. J. Biol. Chem. 1974, 249, 6945–6950. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.F.; et al. CPS1 Maintains Pyrimidine Pools and DNA Synthesis in KRAS/LKB1-Mutant Lung Cancer Cells. Nature 2017, 546, 168–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, B.G.; Wolfe, L.A.; Ichikawa, M.; Markello, T.; He, M.; Tifft, C.J.; Gahl, W.A.; Freeze, H.H. Biallelic Mutations in CAD, Impair de Novo Pyrimidine Biosynthesis and Decrease Glycosylation Precursors. Hum. Mol. Genet. 2015, 24, 3050–3057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, J.; Mayr, J.A.; Alhaddad, B.; Rauscher, C.; Bierau, J.; Kovacs-Nagy, R.; Coene, K.L.; Bader, I.; Holzhacker, M.; Prokisch, H.; et al. CAD Mutations and Uridine-Responsive Epileptic Encephalopathy. Brain 2017, 140, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Caño-Ochoa, F.; Ng, B.G.; Abedalthagafi, M.; Almannai, M.; Cohn, R.D.; Costain, G.; Elpeleg, O.; Houlden, H.; Karimiani, E.G.; Liu, P.; et al. Cell-Based Analysis of CAD Variants Identifies Individuals Likely to Benefit from Uridine Therapy. Genet. Med. 2020, 22, 1598–1605. [Google Scholar] [CrossRef]
- Rymen, D.; Lindhout, M.; Spanou, M.; Ashrafzadeh, F.; Benkel, I.; Betzler, C.; Coubes, C.; Hartmann, H.; Kaplan, J.D.; Ballhausen, D.; et al. Expanding the Clinical and Genetic Spectrum of CAD Deficiency: An Epileptic Encephalopathy Treatable with Uridine Supplementation. Genet. Med. 2020, 22, 1589–1597. [Google Scholar] [CrossRef]
- Lee, L.; Kelly, R.E.; Pastra-Landis, S.C.; Evans, D.R. Oligomeric Structure of the Multifunctional Protein CAD That Initiates Pyrimidine Biosynthesis in Mammalian Cells. Proc. Natl. Acad. Sci. USA 1985, 82, 6802–6806. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Morcillo, M.; Grande-Garcia, A.; Ruiz-Ramos, A.; Del Cano-Ochoa, F.; Boskovic, J.; Ramon-Maiques, S. Structural Insight into the Core of CAD, the Multifunctional Protein Leading De Novo Pyrimidine Biosynthesis. Structure 2017, 25, 912–923.e5. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kelly, R.E.; Evans, D.R. The Structural Organization of the Hamster Multifunctional Protein CAD. Controlled Proteolysis, Domains, and Linkers. J. Biol. Chem. 1992, 267, 7177–7184. [Google Scholar] [CrossRef]
- Mally, M.I.; Grayson, D.R.; Evans, D.R. Controlled Proteolysis of the Multifunctional Protein That Initiates Pyrimidine Biosynthesis in Mammalian Cells: Evidence for Discrete Structural Domains. Proc. Natl. Acad. Sci. USA 1981, 78, 6647–6651. [Google Scholar] [CrossRef]
- Bellin, L.; Del Caño-Ochoa, F.; Velázquez-Campoy, A.; Möhlmann, T.; Ramón-Maiques, S. Mechanisms of Feedback Inhibition and Sequential Firing of Active Sites in Plant Aspartate Transcarbamoylase. Nat. Commun. 2021, 12, 947. [Google Scholar] [CrossRef]
- Lipscomb, W.N.; Kantrowitz, E.R. Structure and Mechanisms of Escherichia Coli Aspartate Transcarbamoylase. Acc. Chem. Res. 2011, 45, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ramos, A.; Velazquez-Campoy, A.; Grande-Garcia, A.; Moreno-Morcillo, M.; Ramon-Maiques, S. Structure and Functional Characterization of Human Aspartate Transcarbamoylase, the Target of the Anti-Tumoral Drug PALA. Structure 2016, 24, 1081–1094. [Google Scholar] [CrossRef] [Green Version]
- Grande-Garcia, A.; Lallous, N.; Diaz-Tejada, C.; Ramon-Maiques, S. Structure, Functional Characterization, and Evolution of the Dihydroorotase Domain of Human CAD. Structure 2014, 22, 185–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Cano-Ochoa, F.; Grande-Garcia, A.; Reverte-Lopez, M.; D’Abramo, M.; Ramon-Maiques, S. Characterization of the Catalytic Flexible Loop in the Dihydroorotase Domain of the Human Multi-Enzymatic Protein CAD. J. Biol. Chem. 2018, 293, 18903–18913. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Davidson, J.N. Substitutions in the Aspartate Transcarbamoylase Domain of Hamster CAD Disrupt Oligomeric Structure. Proc. Natl. Acad. Sci. USA 2000, 97, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Thoden, J.B.; Holden, H.M.; Wesenberg, G.; Raushel, F.M.; Rayment, I. Structure of Carbamoyl Phosphate Synthetase: A Journey of 96 A from Substrate to Product. Biochemistry 1997, 36, 6305–6316. [Google Scholar] [CrossRef] [PubMed]
- de Cima, S.; Polo, L.M.; Diez-Fernandez, C.; Martinez, A.I.; Cervera, J.; Fita, I.; Rubio, V. Structure of Human Carbamoyl Phosphate Synthetase: Deciphering the on/off Switch of Human Ureagenesis. Sci. Rep. 2015, 5, 16950. [Google Scholar] [CrossRef] [Green Version]
- Collins, K.D.; Stark, G.R. Aspartate Transcarbamylase Interaction with the Transition State Analogue N-(Phosphonacetyl)-L-Aspartate. J. Biol. Chem. 1971, 246, 6599–6605. [Google Scholar] [CrossRef]
- Fernandez-Leiro, R.; Scheres, S.H.W. Unravelling Biological Macromolecules with Cryo-Electron Microscopy. Nature 2016, 537, 339–346. [Google Scholar] [CrossRef]
- D’Imprima, E.; Floris, D.; Joppe, M.; Sánchez, R.; Grininger, M.; Kühlbrandt, W. Protein Denaturation at the Air-Water Interface and How to Prevent It. eLife 2019, 8, e42747. [Google Scholar] [CrossRef] [PubMed]
- Rozbeský, D.; Rosůlek, M.; Kukačka, Z.; Chmelík, J.; Man, P.; Novák, P. Impact of Chemical Cross-Linking on Protein Structure and Function. Anal. Chem. 2018, 90, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Thornton, D.T.; Nguyen, H.T. Chemical Cross-Linking in the Structural Analysis of Protein Assemblies. Methods 2018, 144, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Perry, L.J.; Wetzel, R. Disulfide Bond Engineered into T4 Lysozyme: Stabilization of the Protein toward Thermal Inactivation. Science 1984, 226, 555–557. [Google Scholar] [CrossRef]
- Jacobson, R.H.; Matsumura, M.; Faber, H.R.; Matthews, B.W. Structure of a Stabilizing Disulfide Bridge Mutant That Closes the Active-Site Cleft of T4 Lysozyme. Protein. Sci. 1992, 1, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Qu, K.; Chen, Q.; Ciazynska, K.A.; Liu, B.; Zhang, X.; Wang, J.; He, Y.; Guan, J.; He, J.; Liu, T.; et al. Engineered Disulfide Reveals Structural Dynamics of Locked SARS-CoV-2 Spike. PLoS Pathog. 2022, 18, e1010583. [Google Scholar] [CrossRef]
- Qiu, Y.; Davidson, J.N. Aspartate-90 and Arginine-269 of Hamster Aspartate Transcarbamylase Affect the Oligomeric State of a Chimaeric Protein with an Escherichia Coli Maltose-Binding Domain. Biochem. J. 1998, 329 Pt 2, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Berrow, N.S.; Alderton, D.; Sainsbury, S.; Nettleship, J.; Assenberg, R.; Rahman, N.; Stuart, D.I.; Owens, R.J. A Versatile Ligation-Independent Cloning Method Suitable for High-Throughput Expression Screening Applications. Nucleic Acids Res. 2007, 35, e45. [Google Scholar] [CrossRef]
- Ho, S.N.; Hunt, H.D.; Horton, R.M.; Pullen, J.K.; Pease, L.R. Site-Directed Mutagenesis by Overlap Extension Using the Polymerase Chain Reaction. Gene 1989, 77, 51–59. [Google Scholar] [CrossRef]
- Studier, F.W. Protein Production by Auto-Induction in High Density Shaking Cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Vonrhein, C.; Flensburg, C.; Keller, P.; Sharff, A.; Smart, O.; Paciorek, W.; Womack, T.; Bricogne, G. Data Processing and Analysis with the AutoPROC Toolbox. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 293–302. [Google Scholar] [CrossRef] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser Crystallographic Software. J. Appl Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and Development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the Refinement of Macromolecular Crystal Structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Niesen, F.H.; Berglund, H.; Vedadi, M. The Use of Differential Scanning Fluorimetry to Detect Ligand Interactions That Promote Protein Stability. Nat. Protoc. 2007, 2, 2212–2221. [Google Scholar] [CrossRef]
- Prescott, L.M.; Jones, M.E. Modified Methods for the Determination of Carbamyl Aspartate. Anal. Biochem. 1969, 32, 408–419. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Apo-State | Bound to CP | |
|---|---|---|
| Data Collection | ||
| Wavelength (Å) | 0.97926 | 0.97926 |
| Space group | P4332 | P4332 |
| Unit cell: a, b, c (Å) α, β, γ (°) | 139.8, 139.8, 139.8 90, 90, 90 | 139.4, 139.4, 139.4 90, 90, 90 |
| Resolution (Å) | 49.08–2.03 (2.10–2.03) | 49.29–1.58 (1.64–1.58) |
| Reflections (observed/unique) | 325,801/30,158 (30,416/2906) | 900,158/63,482 (78,877/5919) |
| Multiplicity | 10.8 (10.5) | 14.2 (13.3) |
| Rpim | 0.019 (0.43) | 0.021 (0.71) |
| I/σI | 19.9 (1.7) | 19.8 (1.1) |
| Completeness (%) | 100 (100) | 99.7 (96.8) |
| CC1/2 | 1.0 (0.70) | 1.0 (0.45) |
| Wilson B factor (Å2) | 47.67 | 26.57 |
| Refinement | ||
| Resolution (Å) | 49.08–2.03 | 49.29–1.58 |
| Reflections | 30,113 | 63,194 |
| R-factor/Rfree (%) | 17.11/19.99 | 14.58/17.32 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.006 | 0.013 |
| Bond angles (°) | 0.864 | 1.21 |
| N° atoms (no H) | ||
| Protein + ligand | 2594 | 2644 |
| Water | 62 | 291 |
| Ramachandran plot | ||
| Favored (%) | 96.74 | 97.78 |
| Allowed (%) | 3.26 | 2.22 |
| Outliers (%) | 0.00 | 0.00 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
del Caño-Ochoa, F.; Rubio-del-Campo, A.; Ramón-Maiques, S. A Tailored Strategy to Crosslink the Aspartate Transcarbamoylase Domain of the Multienzymatic Protein CAD. Molecules 2023, 28, 660. https://doi.org/10.3390/molecules28020660
del Caño-Ochoa F, Rubio-del-Campo A, Ramón-Maiques S. A Tailored Strategy to Crosslink the Aspartate Transcarbamoylase Domain of the Multienzymatic Protein CAD. Molecules. 2023; 28(2):660. https://doi.org/10.3390/molecules28020660
Chicago/Turabian Styledel Caño-Ochoa, Francisco, Antonio Rubio-del-Campo, and Santiago Ramón-Maiques. 2023. "A Tailored Strategy to Crosslink the Aspartate Transcarbamoylase Domain of the Multienzymatic Protein CAD" Molecules 28, no. 2: 660. https://doi.org/10.3390/molecules28020660