

Mechanistic Aspects of the Electrochemical Oxidation of Aliphatic Amines and Aniline Derivatives


Abstract
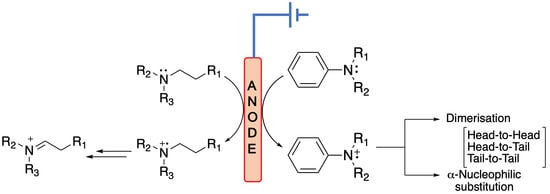
:
1. Introduction
2. Aliphatic Amines














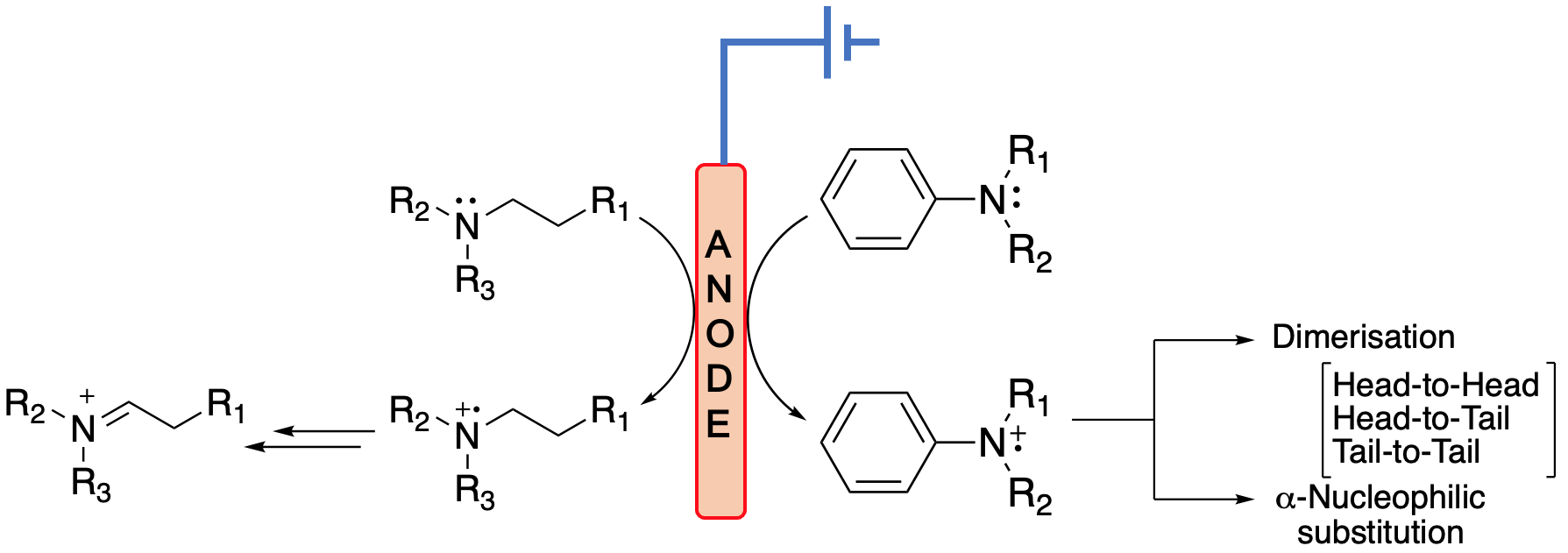{kind=link}
| Entry # | Starting Amine | Nucleophile | Product | % Yield | Ref. |
|---|---|---|---|---|---|
| 1 |  | CH3OH |  | 48 | [37] |
 | 12 | ||||
| 2 |  | Intramolecular OH |  | 25 | [37] |
 | 45 | ||||
| 3 |  | Intramolecular OH |  | 25 | |
 | 45 | ||||
| 4 |  | –CN |  | 53 | [38] |
| DEM– |  | 80 | [39] | ||
| DEP– |  | 70 | |||
| 5 |  | –CN |  | 36 | [38] |
| 6 |  | –CN |  | 31 | [38] |
 | |||||
| 7 |  | –CN |  | 32 | [38] |
 | |||||
| 8 |  | –CN |  | 40 | [38] |
 | |||||
| 9 |  | –CN |  | 40 | [38] |
| 10 |  | –CN |  | 43 | [38] |
| 11 |  | –CN |  | 46 | [38] |
 | |||||
| 12 |  | –CN |  | 57 | [38] |
 | |||||
| 13 |  | –CN |  | 57 | [38] |
 | |||||
| 14 |  | –CN |  | 61 | [38] |
 | |||||
| 15 |  | –CN |  | 59 | [38] |
| 16 |  | –CN |  | 57 | [38] |
| 17 |  | –CN |  | 62 | [38] |
| 18 |  | –CN |  | 63 | [38] |
| 19 |  | –CN |  | – | [40] |
| 20 |  | Intramolecular OH |  | 72 | [41,42] |
| –CH2CN |  | 22 | [42] | ||
| –CN |  | 23 | |||
| 21 |  | DEM– |  | 76 | [39] |
| DEP– |  | 60 | |||
| 22 |  |  |  | 74 | [43] |
| 23 |  | HCOO− |  | 79 | [44] |
| 24 |  |  |  | 90 | [44] |
 |  | 82 | [44] | ||
| 25 |  | H2O |  | 55 | [45] |
| 26 |  | H2O |  | 64 | [45] |
| 27 |  | –CN |  | 70 | [46] |
| 28 |  | –CN |  | 70 | [46] |
| 29 |  | Intramolecular OH |  | 48 | [47] |
| 30 |  | CH3OH |  | 76 | [48] |
| 31 |  | Intramolecular C=C |  | 89 | [49,50] |
| 32 |  | Intramolecular N |  | 79 | [51] |
| 33 |  | CH3OH |  | 88 | [52] |
 |  | 87 | |||
| CH3COOH |  | 54 | |||
| 34 |  | TsN3 |  | 68 | [53] |
| 35 |  | TsN3 |  | 76 | [53] |
| 36 |  | TsN3 |  | 52 | [53] |


2.1. Catalysed Oxidation of Amines


| Entry # | Starting Amine | Second Reactant | Redox Catalyst | Product | % Yield | Ref. |
|---|---|---|---|---|---|---|
| 1 |  | H2O | I− |  | 75 | [65] |
| 2 |  |  | I− |  | 85 | [65] |
| 3 |  |  | I− |  | 83 | [65] |
| 4 |  |  | I− |  | 75 | [65] |
 | I− |  | 17 | [65] | ||
 | I− |  | 79 | [66] | ||
| 5 |  | H2O | I− |  | 53 | [65] |
| 6 |  |  | I− |  | 81 | [66] |
| 7 |  | – | I− |  | 74 | [67] |
| 8 |  | H2O | Br− |  | 50 | [68] |
| 9 |  | H2O | Br− |  | 85 | [68] |
| 10 |  | PhS-SPh | Br− |  | 72 | [69] |
| 11 |  | CH3OH | Cl− |  | 83 | [70] |
| 12 |  | – | Cl− |  | 66 | [67] |
| 13 |  | CH3OH | Cl− |  | 38 | [51] |
| 14 |  | CH3OH | Cl− |  | 65 | [71] |






2.2. Examples of Applications


2.3. Alkanolamines, Amides, Carbamates and Lactams









3. Aniline and Derivatives


- (i).
- Two resonance structures 26 can react through a tail-to-tail coupling to form dimer 27 (Equation (36)).
- (ii).
- The nitrenium cation can react with starting molecule through a head-to-tail coupling to form dimer 28. The same outcome is obtained by reacting 24 with 26 (Equation (37)).
- (iii).
- Two resonance structures 24 can react via a head-to-head coupling to form dimer 30 (Equation (38)).




3.1. Aniline and Para-Substituted Anilines

3.2. N-Substituted Anilines


3.3. N,N-Disubstituted Anilines



3.4. Aminophenols and N-Acylated Anilines





3.5. Catalysed Oxidation of Aniline and Its Derivatives




4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lund, H.; Hammerich, O. Organic Electrochemistry, 4th ed.; Marcel Dekker: New York, NY, USA, 2001. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods: Fundamentals and Applications; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Najmi, A.A.; Bischoff, R.; Permentier, H.P. N-dealkylation of amines. Molecules 2022, 27, 3293. [Google Scholar] [CrossRef]
- Steckhan, E. Anodic oxidation of nitrogen-containing compounds. In Organic Electrochemistry, 4th ed.; Lund, H., Hammerich, O., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Chen, N.; Xu, H.-C. Electrochemical generation of nitrogen-centered radicals for organic synthesis. Green Synth. Catal. 2021, 2, 165–178. [Google Scholar] [CrossRef]
- Kärkäs, M.D. Electrochemical strategies for C–H functionalization and C–N bond formation. Chem. Soc. Rev. 2018, 47, 5786–5865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shono, T. Electroorganic chemistry in organic synthesis. Tetrahedron 1984, 40, 811–850. [Google Scholar] [CrossRef]
- Jaworski, J.S. Organic Electrochemistry: Revised and Expanded; Taylor & Francis Group: Boca Raton, FL, USA, 2015. [Google Scholar]
- Barnes, K.K.; Mann, C.K. Electrochemical oxidation of primary aliphatic amines. J. Org. Chem. 1967, 32, 1474–1479. [Google Scholar] [CrossRef]
- Mann, C.K. Cyclic stationary electrode voltammetry of some aliphatic amines. Anal. Chem. 1964, 36, 2424–2426. [Google Scholar] [CrossRef]
- O’Donnell, J.F.; Mann, C.K. Determination of aliphatic amides by controlled-potential coulometry. J. Electroanal. Chem. Interfacial Electrochem. 1967, 13, 163–166. [Google Scholar] [CrossRef]
- O’Donnell, J.F.; Mann, C.K. Controlled-potential oxidation of aliphatic amides. J. Electroanal. Chem. Interfacial Electrochem. 1967, 13, 157–162. [Google Scholar] [CrossRef]
- Portis, L.C.; Bhat, V.V.; Mann, C.K. Electrochemical dealkylation of aliphatic tertiary and secondary amines. J. Org. Chem. 1970, 35, 2175–2178. [Google Scholar] [CrossRef]
- Smith, P.J.; Mann, C.K. Anodic methoxylation of tertiary amines. J. Org. Chem. 1968, 33, 316–317. [Google Scholar] [CrossRef]
- Smith, P.J.; Mann, C.K. Electrochemical dealkylation of aliphatic amines. J. Org. Chem. 1969, 34, 1821–1826. [Google Scholar] [CrossRef]
- Adenier, A.; Chehimi, M.M.; Gallardo, I.; Pinson, J.; Vila, N. Electrochemical oxidation of aliphatic amines and their attachment to carbon and metal surfaces. Langmuir 2004, 20, 8243–8253. [Google Scholar] [CrossRef]
- Torriero, A.A.J.; Morda, J.; Saw, J. Electrocatalytic dealkylation of amines mediated by ferrocene. Organometallics 2019, 38, 4280–4287. [Google Scholar] [CrossRef]
- Torriero, A.A.J.; Shiddiky, M.J.A.; Burgar, I.; Bond, A.M. Homogeneous electron-transfer reaction between electrochemically generated ferrocenium ions and amine containing compounds. Organometallics 2013, 32, 5731–5739. [Google Scholar] [CrossRef]
- Bock, H.; Goebel, I.; Havlas, Z.; Liedle, S.; Oberhammer, H. Triisopropylamine: A sterically overcrowded molecule with a flattened nc3 pyramid and a “p-type” nitrogen electron pair. Angew. Chem. Int. Ed. Engl. 1991, 30, 187–190. [Google Scholar] [CrossRef]
- Deinhammer, R.S.; Ho, M.; Anderegg, J.W.; Porter, M.D. Electrochemical oxidation of amine-containing compounds: A route to the surface modification of glassy carbon electrodes. Langmuir 1994, 10, 1306–1313. [Google Scholar] [CrossRef]
- Bauer, R.; Wendt, H. Anodic N-N Linkage by oxidation of anions of primary and secondary amines. Angew. Chem. Int. Ed. Engl. 1978, 17, 202–203. [Google Scholar] [CrossRef]
- Loveland, J.W.; Dimeler, G.R. Anodic voltammetry to +2.0 Volts. Application to hydrocarbons and oxidation stability studies. Anal. Chem. 1961, 33, 1196–1201. [Google Scholar] [CrossRef]
- Wawzonek, S.; McIntyre, T.W. Electrolytic oxidation of aromatic amines. J. Electrochem. Soc. 1967, 114, 1025–1029. [Google Scholar] [CrossRef]
- Dvořák, V.; Němec, I.; Zýka, J. Electrochemical oxidation of some aromatic amines in acetonitrile medium. I. N,N-dimethylaniline, triphenylamine, diphenylamine and di-4-tolylamine. Microchem. J. 1967, 12, 99–116. [Google Scholar] [CrossRef]
- Seo, E.T.; Nelson, R.F.; Fritsch, J.M.; Marcoux, L.S.; Leedy, D.W.; Adams, R.N. Anodic oxidation pathways of aromatic amines. Electrochemical and electron paramagnetic resonance studies. J. Am. Chem. Soc. 1966, 88, 3498–3503. [Google Scholar] [CrossRef]
- Yang, H.; Wipf, D.O.; Bard, A.J. Application of rapid scan cyclic voltammetry to a study of the oxidation and dimerization of N,N-dimethylaniline in acetonitrile. J. Electroanal. Chem. 1992, 331, 913–924. [Google Scholar] [CrossRef]
- Zweig, A.; Maurer, A.H.; Roberts, B.G. Oxidation, reduction, and electrochemiluminescence of donor-substituted polycyclic aromatic hydrocarbons. J. Org. Chem. 1967, 32, 1322–1329. [Google Scholar] [CrossRef]
- Zweig, A.; Lancaster, J.E.; Neglia, M.T.; Jura, W.H. Cumulative influence of dimethylamino groups on the π-system properties of aromatic hydrocarbons. J. Am. Chem. Soc. 1964, 86, 4130–4136. [Google Scholar] [CrossRef]
- Novak Jovanović, I.; Jadreško, D.; Miličević, A.; Hranjec, M.; Perin, N. An electrochemical study on the redox chemistry of cyclic benzimidazole derivatives with potent anticancer activity. Electrochim. Acta 2019, 297, 452–462. [Google Scholar] [CrossRef]
- Ross, S.D. The mechanism of anodic dealkylation of aliphatic amines in acetonitrile. Tetrahedron Lett. 1973, 14, 1237–1240. [Google Scholar] [CrossRef]
- Barry, J.E.; Finkelstein, M.; Mayeda, E.A.; Ross, S.D. Products and mechanisms in the anodic oxidation of N,N-dimethylbenzylamine in methanol. J. Org. Chem. 1974, 39, 2695–2699. [Google Scholar] [CrossRef]
- Weinberg, N.L. Anodic oxidation of organic compounds. IV. Mechanism of electrochemical methoxylation of N,N-dimethylbenzylamine. J. Org. Chem. 1968, 33, 4326–4329. [Google Scholar] [CrossRef]
- Becker, J.Y.; Miller, L.L.; Stermitz, F.R. Electrode processes in the oxidation of tetrahydroisoquinoline derivatives. J. Electroanal. Chem. Interfacial Electrochem. 1976, 68, 181–191. [Google Scholar] [CrossRef]
- Lee, K.; Choi, H.; An, J.; Kwon, K.-Y. Stainless steel promoted the electrochemical oxidation of amines into imines. Bull. Korean Chem. Soc. 2022, 43, 937–940. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Ippoliti, J.T. The deprotonation of trialkylamine cation radicals by amines. J. Am. Chem. Soc. 1986, 108, 4879–4881. [Google Scholar] [CrossRef]
- Nelsen, S.F.; Kessel, C.R. 9-butylazabicyclo[3.3.1]nonane radical cation, the first long-lived saturated amine radical cation. J. Chem. Soc. Chem. Commun. 1977, 490–491. [Google Scholar] [CrossRef]
- Weinberg, N.L.; Brown, E.A. The anodic oxidation of organic compounds. II. The electrochemical alkoxylation of tertiary amines. J. Org. Chem. 1966, 31, 4058–4061. [Google Scholar] [CrossRef]
- Chiba, T.; Takata, Y. Anodic cyanation of tertiary aliphatic and heterocyclic amines. J. Org. Chem. 1977, 42, 2973–2977. [Google Scholar] [CrossRef]
- Bidan, G.; Genies, M. Utilisation en synthese du cation iminium genere in situ par oxydation electrochimique d’amines tertiaires. Tetrahedron 1981, 37, 2297–2301. [Google Scholar] [CrossRef]
- Alipour Najmi, A.; Xiao, Z.; Bischoff, R.; Dekker, F.J.; Permentier, H.P. Electrochemical N-demethylation of tropane alkaloids. Green Chem. 2020, 22, 6455–6463. [Google Scholar] [CrossRef]
- Tan, G.-H.; Lim, T.-M.; Kam, T.-S. Electrochemical oxidation of aspidofractinine-type indole alkaloids. A facile, electrochemically-mediated conversion of kopsingine to kopsidines A, B, C, and kopsinganol. Tetrahedron Lett. 1995, 36, 1327–1330. [Google Scholar] [CrossRef]
- Kam, T.-S.; Lim, T.-M.; Tan, G.-H. Electrochemical oxidation of aspidofractinine-type alkaloids: Formation of kopsine, kopsidine, kopsinitarine and bisindole derivatives. J. Chem. Soc. Perkin Trans. 2001, 1, 1594–1604. [Google Scholar] [CrossRef]
- Shono, T.; Matsumura, Y.; Tsubata, K. Electroorganic chemistry. 46. A new carbon-carbon bond forming reaction at the.alpha.-position of amines utilizing anodic oxidation as a key step. J. Am. Chem. Soc. 1981, 103, 1172–1176. [Google Scholar] [CrossRef]
- Ross, S.D.; Finkelstein, M.; Petersen, R.C. Anodic oxidations. III. The reaction mechanism in the electrochemical acetoxylation and alkoxylation of N,N-dimethylamides. J. Am. Chem. Soc. 1966, 88, 4657–4660. [Google Scholar] [CrossRef]
- Okita, M.; Wakamatsu, T.; Ban, Y. Anodic oxidation of N-alkyl-lactams. J. Chem. Soc. Chem. Commun. 1979, 749. [Google Scholar] [CrossRef]
- Asher, V.; Becu, C.; Anteunis, M.J.O.; Callens, R. New synthesis of pipecolic acid and analogs. Tetrahedron Lett. 1981, 22, 141–144. [Google Scholar] [CrossRef]
- Cornille, F.; Slomczynska, U.; Smythe, M.L.; Beusen, D.D.; Moeller, K.D.; Marshall, G.R. Electrochemical cyclization of dipeptides toward novel bicyclic, reverse-turn peptidomimetics. 1. Synthesis and conformational analysis of 7,5-bicyclic systems. J. Am. Chem. Soc. 1995, 117, 909–917. [Google Scholar] [CrossRef]
- Danielmeier, K.; Schierle, K.; Steckhan, E. A new chiral, cationic β-amino alcohol equivalent: A variable approach to enantiomerically pure building blocks for hydroxyethylene isosters. Angew. Chem. Int. Ed. Engl. 1996, 35, 2247–2248. [Google Scholar] [CrossRef]
- Li, W.; Hanau, C.E.; dAvignon, A.; Moeller, K.D. Conformationally restricted peptide mimetics: The incorporation of 6,5-bicyclic lactam ring skeletons into peptides. J. Org. Chem. 1995, 60, 8155–8170. [Google Scholar] [CrossRef]
- Moeller, K.D.; Rothfus, S.L.; Wong, P.L. Anodic amide oxidations: Fundamental studies concerning the annulation of six- and seven-membered rings onto amines. Tetrahedron 1991, 47, 583–592. [Google Scholar] [CrossRef]
- Papadopoulos, A.; Lewall, B.; Steckhan, E.; Ginzel, K.-D.; Knoch, F.; Nieger, M. Anodic oxidation of N-acyl and N-alkoxycarbonyl dipeptide esters as a key step for the formation of chiral heterocyclic synthetic building blocks. Tetrahedron 1991, 47, 563–572. [Google Scholar] [CrossRef]
- Rudd, E.J.; Finkelstein, M.; Ross, S.D. Anodic oxidations. VII. Reaction mechanism in the electrochemical oxidation of N,N-dimethylformamide in acetic acid and in methanol. J. Org. Chem. 1972, 37, 1763–1767. [Google Scholar] [CrossRef]
- Zhang, L.; Su, J.-H.; Wang, S.; Wan, C.; Zha, Z.; Du, J.; Wang, Z. Direct electrochemical imidation of aliphatic amines via anodic oxidation. Chem. Commun. 2011, 47, 5488–5490. [Google Scholar] [CrossRef]
- Gad, S.C. Ethyleneimine. In Encyclopedia of Toxicology (Third Edition); Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 532–534. [Google Scholar]
- Gassman, P.G.; Nishiguchi, I.; Yamamoto, H. Anodic oxidation of aziridines. J. Am. Chem. Soc. 1975, 97, 1600–1602. [Google Scholar] [CrossRef]
- Torriero, A.A.J. Electrochemistry in Ionic Liquids. Volume 1: Fundamentals; Springer: Cham, Switzerland, 2015. [Google Scholar]
- Torriero, A.A.J.; Feldberg, S.; Zhang, J.; Simonov, A.; Bond, A. On choosing a reference redox system for electrochemical measurements: A cautionary tale. J. Solid State Electrochem. 2013, 17, 3021–3026. [Google Scholar] [CrossRef]
- Gao, Z.-N.; Zhang, J.; Liu, W.-Y. Electrocatalytic oxidation of N-acetyl-L-cysteine by acetylferrocene at glassy carbon electrode. Electroanal. Chem. Interfacial Electrochem. 2005, 580, 9–16. [Google Scholar] [CrossRef]
- Largeron, M.; Neudorffer, A.; Benoit, M.; Fleury, M.-B. Oxidation of primary aliphatic amines catalyzed by an electrogenerated quinonoid species: A small molecule mimic of amine oxidases. Proc. Electrochem. Soc. 2003, 12, 185–188. [Google Scholar]
- Shiddiky, M.J.A.; Torriero, A.A.J.; Zeng, Z.; Spiccia, L.; Bond, A.M. Highly selective and sensitive DNA assay based on electrocatalytic oxidation of ferrocene bearing zinc(ii)-cyclen complexes with diethylamine. J. Am. Chem. Soc. 2010, 132, 10053–10063. [Google Scholar] [CrossRef] [PubMed]
- Togni, A.; Hayashi, T. Ferrocenes: Homogeneous Catalysis, Organic Synthesis, Materials Science; Wiley: New York, NY, USA, 2008. [Google Scholar]
- Fleischmann, M.; Korinek, K.; Pletcher, D. The oxidation of organic compounds at a nickel anode in alkaline solution. J. Electroanal. Chem. Interfacial Electrochem. 1971, 31, 39–49. [Google Scholar] [CrossRef]
- Torii, S.; Inokuchi, T.; Takagishi, S.; Sato, E.; Tsujiyama, H. Conversion of cyclic aziridines and 2-amino-1-cycloalkanols to keto nitriles by using electrogenerated reactive halogen species. Chem. Lett. 1987, 16, 1469–1472. [Google Scholar] [CrossRef]
- Bishop, R. 6.07 Ritter-type reactions. In Comprehensive Organic Synthesis, 2nd ed.; Knochel, P., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; pp. 239–295. [Google Scholar]
- Shono, T.; Matsumura, Y.; Hayashi, J.; Usui, M.; Yamane, S.-I.; Inoue, K. Electroorganic chemistry. 66. Electrochemical oxidation of aminals and enamines using a mediatory system. Acta Chem. Scand. 1983, B37, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Torii, S.; Sayo, N.; Tanaka, H. Electrosynthesis of heteroatom bonds. 5. Direct cross-coupling of dialkylphosphites with amines by an iodonium ion-promoted electrolytic procedure. Tetrahedron Lett. 1979, 20, 4471–4474. [Google Scholar] [CrossRef]
- Okimoto, M.; Takahashi, Y.; Numata, K.; Nagata, Y.; Sasaki, G. Electrochemical oxidation of benzylic amines into the corresponding imines in the presence of catalytic amounts of KI. Synth. Commun. 2005, 35, 1989–1995. [Google Scholar] [CrossRef]
- Billon-Souquet, F.; Martens, T.; Royer, J. Anodic oxidation of the N-cyanomethyloxazolidine system. Tetrahedron 1996, 52, 15127–15136. [Google Scholar] [CrossRef]
- Torii, S.; Tanaka, H.; Hamano, S.-i.; Tada, N.; Nokami, J.; Sasaoka, M. MgBr2-promoted electrosynthesis of sulfenimines (thiooximes) from α-aminoalkanoates and diaryl/dialkyl disulfides in a CH2Cl2–H2O two-phase system. a straightforward preparation of C(6)/C(7)-sulfenimine derivatives of penicillin and cephalosporin. Chem. Lett. 1984, 13, 1823–1826. [Google Scholar] [CrossRef]
- Shono, T.; Matsumura, Y.; Inoue, K. Electroorganic chemistry 71. Anodic alpha-methoxylation of N-carbomethoxylated or N-acylated alpha-amino acid esters and alpha-amino-beta-lactams. J. Org. Chem. 1983, 48, 1388–1389. [Google Scholar] [CrossRef]
- Papadopoulos, A.; Heyer, J.; Ginzel, K.-D.; Steckhan, E. Regioselective anodic oxidation of N-acyl, N-alkoxycarbonyl, and N-(2-Nitrophenylsulfenyl) dipeptide esters. Chem. Ber. 1989, 122, 2159–2164. [Google Scholar] [CrossRef]
- Cãproiu, M.; Florea, C.; Galli, C.; Petride, A.; Petride, H. Oxidation of n-benzyl aziridine by molecular iodine: Competition of electron transfer and heterolytic pathways. Eur. J. Org. Chem. 2000, 2000, 1037–1043. [Google Scholar] [CrossRef]
- Kossaï, R.; Simonet, J.; Dauphin, G. The anodic tetramerization of the N-benzylaziridine: A chain process. Tetrahedron Lett. 1980, 21, 3575–3578. [Google Scholar] [CrossRef]
- Cuppoletti, A.; Dagostin, C.; Florea, C.; Galli, C.; Gentili, P.; Lanzalunga, O.; Petride, A.; Petride, H. The oxidation of n-benzylaziridine catalyzed by iron porphyrin: Radical versus electron transfer mechanism. Chem.–A Eur. J. 1999, 5, 2993–2999. [Google Scholar] [CrossRef]
- Largeron, M.; Chiaroni, A.; Fleury, M.-B. Environmentally friendly chemoselective oxidation of primary aliphatic amines by using a biomimetic electrocatalytic system. Chem.–A Eur. J. 2008, 14, 996–1003. [Google Scholar] [CrossRef]
- Dapperheld, S.; Steckhan, E.; Brinkhaus, K.-H.G.; Esch, T. Organic electron transfer systems, ii substituted triarylamine cation-radical redox systems—Synthesis, electrochemical and spectroscopic properties, Hammet behavior, and suitability as redox catalysts. Chem. Ber. 1991, 124, 2557–2567. [Google Scholar] [CrossRef]
- Schmidt, W.; Steckhan, E. Über organische elektronenüberträgersysteme, I. Elektrochemische und spektroskopische untersuchung bromsubstituierter triarylamin-redoxsysteme. Chem. Ber. 1980, 113, 577–585. [Google Scholar] [CrossRef]
- Steckhan, E. Indirect electroorganic syntheses—A modern chapter of organic electrochemistry. Angew. Chem. Int. Ed. Engl. 1986, 25, 683–701. [Google Scholar] [CrossRef]
- Heyer, J.; Dapperheld, S.; Steckhan, E. Convenient syntheses of 2-nitrophenylsulfen-(NPS-)imines by oxidation of NPS-protected amines and amino acid derivatives. Chem. Ber. 1988, 121, 1617–1623. [Google Scholar] [CrossRef]
- Pletcher, D.; Zappi, G.D. The indirect anodic oxidation of amines mediated by brominated aryl amines. J. Electroanal. Chem. Interfacial Electrochem. 1989, 265, 203–213. [Google Scholar] [CrossRef]
- Diamond, S.E.; Taube, H. Nucleophilic attack on cyanoformate induced by co-ordination to ruthenium ammines. J. Chem. Soc. Chem. Commun. 1974, 622–623. [Google Scholar] [CrossRef]
- Diamond, S.E.; Tom, G.M.; Taube, H. Ruthenium promoted oxidation of amines. J. Am. Chem. Soc. 1975, 97, 2661–2664. [Google Scholar] [CrossRef]
- Kwok, K.K.; Vincent, E.C.; Gibson, J.N. 36—Antineoplastic drugs. In Pharmacology and Therapeutics for Dentistry, 7th ed.; Dowd, F.J., Johnson, B.S., Mariotti, A.J., Eds.; Mosby: Maryland Heights, MI, USA, 2017; pp. 530–562. [Google Scholar]
- Seneca, Chapter 2-alkaloid chemistry. In Alkaloids-Secrets of Life; Aniszewski, T. (Ed.) Elsevier: Amsterdam, The Netherlands, 2007; pp. 61–139. [Google Scholar]
- Tabakovic, I.; Gunic, E.; Gasic, M.J. Anodic C16–C21 fragmentation of catharanthine in methanol. Synthesis of 16-methoxycleavamine. J. Chem. Soc. Perkin Trans. 1996, 2, 2741–2745. [Google Scholar] [CrossRef]
- Najmi, A.A.; Bhat, M.F.; Bischoff, R.; Poelarends, G.J.; Permentier, H.P. TEMPO-mediated electrochemical N-demethylation of opiate alkaloids. ChemElectroChem 2021, 8, 2590–2596. [Google Scholar] [CrossRef]
- Shono, T.; Toda, T.; Oshino, N. Electron transfer from nitrogen in microsomal oxidation of amine and amide. Simulation of microsomal oxidation by anodic oxidation. J. Am. Chem. Soc. 1982, 104, 2639–2641. [Google Scholar] [CrossRef]
- Shono, T. Synthesis of Alkaloidal Compounds Using an Electrochemical Reaction as a Key Step; Stekchan, E., Ed.; Electrochemistry III: Berlin/Heidelberg, Germany; Springer: Berlin/Heidelberg, Germany, 1988; pp. 131–151. [Google Scholar]
- Portis, L.C.; Klug, J.T.; Mann, C.K. Electrochemical oxidation of some phenethylamines. J. Org. Chem. 1974, 39, 3488–3494. [Google Scholar] [CrossRef]
- Masui, M.; Kamada, Y.; Ozaki, S. Anodic oxidation of amines. VI. Revised reaction scheme for the oxidation of ephedrine. Chem. Pharm. Bull. 1980, 28, 1619–1622. [Google Scholar] [CrossRef] [Green Version]
- Masui, M.; Kamada, Y.; Sasaki, E.; Ozaki, S. Anodic oxidation of amines. VII. Oxidation of beta-alkanolamines in aqueous buffer of pH 10. Chem. Pharm. Bull. 1982, 30, 1234–1243. [Google Scholar] [CrossRef] [Green Version]
- Shono, T.; Hamaguchi, H.; Matsumura, Y.; Yoshida, K. Syntheses of carbonyl compounds by the anodic cleavage of glycols and related compounds. Tetrahedron Lett. 1977, 18, 3625–3628. [Google Scholar] [CrossRef]
- Célimène, C.; Dhimane, H.; Le Bail, M.; Lhommet, G. A short and highly stereocontrolled total synthesis of (3R,5R,8aR)-3-n-butyl-5-methylindolizidine. Tetrahedron Lett. 1994, 35, 6105–6106. [Google Scholar] [CrossRef]
- Beal, L.M.; Moeller, K.D. A sequential electrochemical oxidation—olefin metathesis strategy for the construction of bicyclic lactam based peptidomimetics. Tetrahedron Lett. 1998, 39, 4639–4642. [Google Scholar] [CrossRef]
- Finkelstein, M.; Ross, S.D. Anodic oxidation of N-methylformamide and N-methylacetamide. Tetrahedron 1972, 28, 4497–4502. [Google Scholar] [CrossRef]
- Kardassis, G.; Brungs, P.; Nothhelfer, C.; Steckhan, E. Electrogenerated chiral cationic glycine equivalents—Part 2: Chiral 3-methoxy-2,5-morpholinediones from (S)-α-hydroxy acids and dimethyl aminomalonate. Tetrahedron 1998, 54, 3479–3488. [Google Scholar] [CrossRef]
- Kardassis, G.; Brungs, P.; Steckhan, E. Electrogenerated chiral cationic glycine equivalents—Part 1: The 6-methoxy derivative of cyclo(L-Pro-Gly). Tetrahedron 1998, 54, 3471–3478. [Google Scholar] [CrossRef]
- Moeller, K.D.; Rutledge, L.D. Anodic amide oxidations: The synthesis of two spirocyclic L-pyroglutamide building blocks. J. Org. Chem. 1992, 57, 6360–6363. [Google Scholar] [CrossRef]
- Moeller, K.D.; Sharif, T.; Mohammad, R.M. Electrochemical amide oxidations in the presence of monomethoxylated phenyl rings. An unexpected relationship between the chemoselectivity of the oxidation and the location of the methoxy substituent. Tetrahedron Lett. 1989, 30, 1213–1216. [Google Scholar] [CrossRef]
- Moeller, K.D.; Wang, P.W.; Tarazi, S.; Marzabadi, M.R.; Wong, P.L. Anodic amide oxidations in the presence of electron-rich phenyl rings: Evidence for an intramolecular electron-transfer mechanism. J. Org. Chem. 1991, 56, 1058–1067. [Google Scholar] [CrossRef]
- Schierle, K.; Vahle, R.; Steckhan, E. Efficient pathways to precursors of pyrrolidine azasugar structures. Eur. J. Org. Chem. 1998, 1998, 509–514. [Google Scholar] [CrossRef]
- Skrinjar, M.; Wistrand, L.-G. Highly stereoselective addition of alkylcopper reagents to n-acyliminium ions. Enantioselective synthesis of trans-2- butyl-5-heptylpyrrolidine. Tetrahedron Lett. 1990, 31, 1775–1778. [Google Scholar] [CrossRef]
- Slomczynska, U.; Chalmers, D.K.; Cornille, F.; Smythe, M.L.; Beusen, D.D.; Moeller, K.D.; Marshall, G.R. Electrochemical Cyclization of Dipeptides to Form Novel Bicyclic, Reverse-Turn Peptidomimetics. 2. Synthesis and Conformational Analysis of 6,5-Bicyclic Systems. J. Org. Chem. 1996, 61, 1198–1204. [Google Scholar] [CrossRef]
- Thaning, M.; Wistrand, L.-G. Synthetic applications of electrochemically produced α-methoxyamides. Part 2. Oxidation of hydroxyproline derivatives. Helv. Chim. Acta 1986, 69, 1711–1717. [Google Scholar] [CrossRef]
- Thaning, M.; Wistrand, L.G. Synthetic applications of electrochemically produced .alpha.-methoxy amides. 5. A short enantiodivergent synthesis of the Geissman-Waiss lactone. J. Org. Chem. 1990, 55, 1406–1408. [Google Scholar] [CrossRef]
- Wistrand, L.-G.; Skrinjar, M. Chirospecific synthesis of trans-2,5-disubstituted pyrrolidines via stereoselective addition of organocopper reagents to N-acyliminium ions. Tetrahedron 1991, 47, 573–582. [Google Scholar] [CrossRef]
- Wong, P.L.; Moeller, K.D. Anodic amide oxidations: Total syntheses of (-)-A58365A and (+-)-A58365B. J. Am. Chem. Soc. 1993, 115, 11434–11445. [Google Scholar] [CrossRef]
- Xu, Z.; Huang, Z.; Li, Y.; Kuniyil, R.; Zhang, C.; Ackermann, L.; Ruan, Z. Catalyst-free, direct electrochemical synthesis of annulated medium-sized lactams through C–C bond cleavage. Green Chem. 2020, 22, 1099–1104. [Google Scholar] [CrossRef]
- Yao, H.; Sherer, E.C.; Lu, M.; Small, J.; Martin, G.E.; Lam, Y.-h.; Chen, Q.; Helmy, R.; Liu, Y.; Chen, H. One-step regio- and stereoselective electrochemical synthesis of orexin receptor antagonist oxidative metabolites. J. Org. Chem. 2022, 87, 15011–15021. [Google Scholar] [CrossRef]
- Mikami, R.; Nakamura, Y.; Shida, N.; Atobe, M. Anodic substitution reaction of carbamates in a flow microreactor using a stable emulsion solution. React. Chem. Eng. 2021, 6, 2024–2028. [Google Scholar] [CrossRef]
- Novaes, L.F.T.; Ho, J.S.K.; Mao, K.; Liu, K.; Tanwar, M.; Neurock, M.; Villemure, E.; Terrett, J.A.; Lin, S. Exploring electrochemical C(sp3)–H oxidation for the late-stage methylation of complex molecules. J. Am. Chem. Soc. 2022, 144, 1187–1197. [Google Scholar] [CrossRef]
- Shono, T.; Hamaguchi, H.; Matsumura, Y. Electroorganic chemistry. XX. Anodic oxidation of carbamates. J. Am. Chem. Soc. 1975, 97, 4264–4268. [Google Scholar] [CrossRef]
- Shono, T.; Matsumura, Y.; Onomura, O.; Yamada, Y. A new method for regioselective synthesis of 2-substituted 1-(methoxycarbonyl)-1,2-dihydropyridines. Tetrahedron Lett. 1987, 28, 4073–4074. [Google Scholar] [CrossRef]
- Shono, T.; Matsumura, Y.; Tsubata, K.; Sugihara, Y. A new method of acylation at β-position of aliphatic amines. Tetrahedron Lett. 1982, 23, 1201–1204. [Google Scholar] [CrossRef]
- Shono, T.; Matsumura, Y.; Tsubata, K.; Sugihara, Y.; Yamane, S.; Kanazawa, T.; Aoki, T. Electroorganic chemistry. 60. Electroorganic synthesis of enamides and enecarbamates and their utilization in organic synthesis. J. Am. Chem. Soc. 1982, 104, 6697–6703. [Google Scholar] [CrossRef]
- Shono, T.; Terauchi, S.; Ohki, Y.; Matsumura, Y. Regioselective introduction of alkyl groups to the position α or γ to nitrogen atom of piperidine skeletons using anodic oxidation in a key step. Tetrahedron Lett. 1990, 31, 6385–6386. [Google Scholar] [CrossRef]
- Torii, S.; Inokuchi, T.; Kubota, M. A practical access to methyl 3,3-dimethoxypropionates and N-protected beta-aminoacrylates and beta-aminoacrylonitrile by using electrochemical procedure. J. Org. Chem. 1985, 50, 4157–4160. [Google Scholar] [CrossRef]
- Martínez, Á.M.; Hayrapetyan, D.; van Lingen, T.; Dyga, M.; Gooßen, L.J. Taking electrodecarboxylative etherification beyond Hofer–Moest using a radical C–O coupling strategy. Nat. Commun. 2020, 11, 4407. [Google Scholar] [CrossRef]
- Fuchigami, T.; Iwaoka, T.; Nonaka, T.; Sekine, T. Electrochemical oxidation of 3,3,7,7-tetraethyl-perhydro-1,5-diazocine. Chem. Lett. 1978, 7, 1437–1440. [Google Scholar] [CrossRef] [Green Version]
- Fuchigami, T.; Iwaoka, T.; Nonaka, T.; Sekine, T. Electrochemical generation of unstable nitrogen species. Part 4. Electrochemical and chemical oxidation of cyclic and open-chain diamines. The formation of cyclic hydrazine derivatives. Bull. Chem. Soc. Jpn. 1980, 53, 2040–2045. [Google Scholar] [CrossRef]
- Laube, B.L.; Asirvatham, M.R.; Mann, C.K. Electrochemical oxidation of tropanes. J. Org. Chem. 1977, 42, 670–674. [Google Scholar] [CrossRef]
- Fuchigami, T.; Sato, T.; Nonaka, T. Electrochemical generation of reactive nitrogen species. 10. Anodic amination of tetrahydrofuran. J. Org. Chem. 1986, 51, 366–369. [Google Scholar] [CrossRef]
- Frankowski, K.J.; Liu, R.; Milligan, G.L.; Moeller, K.D.; Aubé, J. Practical electrochemical anodic oxidation of polycyclic lactams for late stage functionalization. Angew. Chem. Int. Ed. 2015, 54, 10555–10558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, M.; Hara, S.; Ozaki, S. Anodic oxidation of amides and lactams using N-hydroxyphthalimide as a mediator. Chem. Pharm. Bull. 1986, 34, 975–979. [Google Scholar] [CrossRef] [Green Version]
- Warning, K.; Mitzlaff, M. Verfahren zur herstellung von ω,ω-dialkoxycarbonsaeureestern und -amiden. Tetrahedron Lett. 1979, 20, 1563–1564. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kuriyama, M.; Onomura, O. Shono-type oxidation for functionalization of N-heterocycles. Chem. Rec. 2021, 21, 2239–2253. [Google Scholar] [CrossRef] [PubMed]
- Okita, M.; Mori, M.; Wakamatsu, T.; Ban, Y. Anodic oxidation of N-alkyl-β-lactams. Heterocycles 1985, 23, 247–250. [Google Scholar] [CrossRef]
- Mori, M.; Kagechika, K.; Sasai, H.; Shibasaki, M. New synthesis of 4-acetoxy-2-azetidinones by use of electrochemical oxidation. Tetrahedron 1991, 47, 531–540. [Google Scholar] [CrossRef]
- Weinberg, N.L.; Weinberg, H.R. Electrochemical oxidation of organic compounds. Chem. Rev. 1968, 68, 449–523. [Google Scholar] [CrossRef]
- Genies, E.M.; Lapkowski, M. Spectroelectrochemical evidence for an intermediate in the electropolymerization of aniline. J. Electroanal. Chem. Interfacial Electrochem. 1987, 236, 189–197. [Google Scholar] [CrossRef]
- Adams, R.N. Anodic oxidation pathways of aromatic hydrocarbons and amines. Acc. Chem. Res. 1969, 2, 175–180. [Google Scholar] [CrossRef]
- Hand, R.L.; Nelson, R.F. The anodic decomposition pathways of ortho- and meta-substituted anilines. J. Electrochem. Soc. 1978, 125, 1059–1069. [Google Scholar] [CrossRef]
- Hand, R.L.; Nelson, R.F. Anodic oxidation pathways of N-alkylanilines. J. Am. Chem. Soc. 1974, 96, 850–860. [Google Scholar] [CrossRef]
- Sharma, L.R.; Manchanda, A.K.; Singh, G.; Verma, R.S. Cyclic voltammetry of aromatic amines in aqueous and non-aqueous media. Electrochim. Acta 1982, 27, 223–233. [Google Scholar] [CrossRef]
- Wawzonek, S.; McIntyre, T.W. Electrolytic preparation of azobenzenes. J. Electrochem. Soc. 1972, 119, 1350. [Google Scholar] [CrossRef]
- Matsuda, Y.; Shono, A.; Iwakura, C.; Ohshiro, Y.; Agawa, T.; Tamura, H. Anodic oxidation of aniline in aqueous alkaline solution. Bull. Chem. Soc. Jpn. 1971, 44, 2960–2963. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Bard, A.J. The application of rapid scan cyclic voltammetry and digital simulation to the study of the mechanism of diphenylamine oxidation, radical cation dimerization, and polymerization in acetonitrile. J. Electroanal. Chem. Interfacial Electrochem. 1991, 306, 87–109. [Google Scholar] [CrossRef]
- Andrieux, C.P.; Gallardo, I.; Junca, M. Mechanistic study of the electrochemical oxidation of some aromatic amines in the presence of bases. J. Electroanal. Chem. 1993, 354, 231–241. [Google Scholar] [CrossRef]
- Nishiyama, S.; Iida, Y.; Hino, S.; Yamamura, S. Anodic oxidation of N-alkylanilines. Electrochim. Acta 1997, 42, 1943–1949. [Google Scholar] [CrossRef]
- Yoshida, K.; Fueno, T. Anodic oxidations. VI. p-cyanation of diphenylamines. J. Org. Chem. 1972, 37, 4145–4147. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Adams, R.N. Anodic oxidation studies of N,N-dimethylaniline. I. Voltammetric and spectroscopic investigations at platinum electrodes. J. Am. Chem. Soc. 1962, 84, 2058–2061. [Google Scholar] [CrossRef]
- Galus, Z.; Adams, R.N. Anodic oxidation studies of N,N-dimethylaniline. II. Stationary and rotated disk studies at inert electrodes. J. Am. Chem. Soc. 1962, 84, 2061–2065. [Google Scholar] [CrossRef]
- Galus, Z.; White, R.M.; Rowland, F.S.; Adams, R.N. Anodic oxidation studies of N,N-dimethylaniline. III. Tritium tracer studies of electrolysis products. J. Am. Chem. Soc. 1962, 84, 2065–2068. [Google Scholar] [CrossRef]
- Martins, D.; Garrido, E.M.P.J.; Borges, F.; Garrido, J.M.P.J. Voltammetric profiling of new psychoactive substances: Piperazine derivatives. J. Electroanal. Chem. 2021, 883, 115054. [Google Scholar] [CrossRef]
- Liu, K.; Tang, S.; Wu, T.; Wang, S.; Zou, M.; Cong, H.; Lei, A. Electrooxidative para-selective C–H/N–H cross-coupling with hydrogen evolution to synthesize triarylamine derivatives. Nat. Commun. 2019, 10, 639. [Google Scholar] [CrossRef]
- Nikl, J.; Ravelli, D.; Schollmeyer, D.; Waldvogel, S.R. Straightforward electrochemical sulfonylation of arenes and aniline derivatives using sodium sulfinates. ChemElectroChem 2019, 6, 4450–4455. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Fang, Y.; Tang, R.; Xu, D.; Dai, S.; Zhang, W. Electrochemical oxidative sulfonylation of N-arylamides/amine with sodium sulfinates. Asian J. Org. Chem. 2022, 11, e202100805. [Google Scholar] [CrossRef]
- Hand, R.; Melicharek, M.; Scoggin, D.; Stotz, R.; Carpenter, A.; Nelson, R. Electrochemical oxidation pathways of substituted dimethylanilines. Collect. Czechoslov. Chem. Commun. 1971, 36, 842–854. [Google Scholar] [CrossRef]
- Marcus, M.F.; Hawley, M.D. The electrochemical oxidation of p-dimethylaminophenol in aqueous solution. J. Electroanal. Chem. Interfacial Electrochem. 1968, 18, 175–183. [Google Scholar] [CrossRef]
- Melicharek, M.; Nelson, R.F. The electrochemical oxidation of N,N-dimethyl-p-toluidine. J. Electroanal. Chem. Interfacial Electrochem. 1970, 26, 201–209. [Google Scholar] [CrossRef]
- Rees, N.V.; Klymenko, O.V.; Compton, R.G.; Oyama, M. The electro-oxidation of N,N-dimethyl-p-toluidine in acetonitrile:: A microdisk voltammetry study. J. Electroanal. Chem. 2002, 531, 33–42. [Google Scholar] [CrossRef]
- Weinberg, N.L.; Reddy, T.B. Anodic oxidation of organic compounds. III. Effect of electrolyte on electrochemical methoxylation and dimerization of N,N-dimethylaniline. J. Am. Chem. Soc. 1968, 90, 91–94. [Google Scholar] [CrossRef]
- Shono, T.; Matsumura, Y.; Inoue, K.; Ohmizu, H.; Kashimura, S. Electroorganic chemistry. 62. Reaction of iminium ion with nucleophile: A versatile synthesis of tetrahydroquinolines and julolidines. J. Am. Chem. Soc. 1982, 104, 5753–5757. [Google Scholar] [CrossRef]
- Andreades, S.; Zahnow, E.W. Anodic cyanations of aromatic compounds. J. Am. Chem. Soc. 1969, 91, 4181–4190. [Google Scholar] [CrossRef]
- Michel, S.; Le Gall, E.; Hurvois, J.-P.; Moinet, C.; Tallec, A.; Uriac, P.; Toupet, L. Anodic cyanation of N-substituted 1-benzazepines: Synthesis of the corresponding α-aminonitriles. Liebigs Ann. 1997, 1997, 259–267. [Google Scholar] [CrossRef]
- Snead, W.K.; Remick, A.E. Studies on oxidation-reduction mechanism. II. The anodic oxidation of p-aminophenol. J. Am. Chem. Soc. 1957, 79, 6121–6127. [Google Scholar] [CrossRef]
- Hawley, D.; Adams, R.N. Homogeneous chemical reactions in electrode processes: Measurement of rates of follow-up chemical reactions. J. Electroanal. Chem. (1959) 1965, 10, 376–386. [Google Scholar] [CrossRef]
- Jackowska, K.; Bukowska, J.; Kudelski, A. Electro-oxidation of o-aminophenol studied by cyclic voltammetry and surface enhanced Raman scattering (SERS). J. Electroanal. Chem. 1993, 350, 177–187. [Google Scholar] [CrossRef]
- Amani, A.; Nematollahi, D. Electrochemical synthesis based on the oxidation of 1-(4-(4-hydroxyphenyl)piperazin-1-yl)ethanone in the presence of nucleophiles. J. Org. Chem. 2012, 77, 11302–11306. [Google Scholar] [CrossRef]
- Breising, V.M.; Kayser, J.M.; Kehl, A.; Schollmeyer, D.; Liermann, J.C.; Waldvogel, S.R. Electrochemical formation of N,N′-diarylhydrazines by dehydrogenative N–N homocoupling reaction. Chem. Commun. 2020, 56, 4348–4351. [Google Scholar] [CrossRef]
- Schulz, L.; Enders, M.; Elsler, B.; Schollmeyer, D.; Dyballa, K.M.; Franke, R.; Waldvogel, S.R. Reagent- and metal-free anodic C−C cross-coupling of aniline derivatives. Angew. Chem. Int. Ed. 2017, 56, 4877–4881. [Google Scholar] [CrossRef]
- Schulz, L.; Franke, R.; Waldvogel, S.R. Direct anodic dehydrogenative cross- and homo-coupling of formanilides. ChemElectroChem 2018, 5, 2069–2072. [Google Scholar] [CrossRef]
- Schulz, L.; Husmann, J.-Å.; Waldvogel, S.R. Outstandingly robust anodic dehydrogenative aniline coupling reaction. Electrochim. Acta 2020, 337, 135786. [Google Scholar] [CrossRef]
- Xiao, H.-L.; Yang, C.-W.; Zhang, N.-T.; Zeng, C.-C.; Hu, L.-M.; Tian, H.-Y.; Little, R.D. Electrochemical oxidation of aminophenols in the presence of benzenesulfinate. Tetrahedron 2013, 69, 658–663. [Google Scholar] [CrossRef]
- Kashiwagi, Y.; Anzai, J.-i. Selective electrocatalytic oxidation of N-alkyl-N-methylanilines to N-akylformanilides using nitroxyl radical. Chem. Pharm. Bull. 2001, 49, 324–326. [Google Scholar] [CrossRef] [Green Version]
- Hunter, D.H.; Racok, J.S.; Rey, A.W.; Zea-Ponce, Y. Oxoiminium ions for N-demethylation: 1-oxo-2,2,6,6-tetramethylpiperidinium chloride. J. Org. Chem. 1988, 53, 1278–1281. [Google Scholar] [CrossRef]
| Amine | Solvent | Supporting Electrolyte | Working Electrode | Eox a (V vs. SCE) | Ref. |
|---|---|---|---|---|---|
| propylamine | CH3CN | 0.1 M Na[ClO4] | Pt | 1.38 | [10] |
| butylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.36 | [16] |
| EtOH | 0.1 M LiClO4 | GC | 1.22 | [20] | |
| THF | 0.1 M LiClO4 | GC | 1.15 | [21] | |
| pentylamine | CH3CN | 0.1 M Na[ClO4] | Pt | 1.45 | [10] |
| hexylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.36 | [16] |
| nonylamine | CH3CN | 0.1 M Na[ClO4] | Pt | 1.48 | [10] |
| t-butylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.44 | [16] |
| THF | 0.1 M LiClO4 | GC | 1.21 | [21] | |
| CH3CN | 0.1 M Na[ClO4] | Pt | 1.40 | [10] | |
| butylamide | THF | 0.1 M LiClO4 | GC | 0.16 | [21] |
| t-butylamide | THF | 0.1 M LiClO4 | GC | −0.10 | [21] |
| cyclohexylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.39 | [16] |
| THF | 0.1 M LiClO4 | GC | 1.26 | [21] | |
| cyclohexylamide | THF | 0.1 M LiClO4 | GC | 0.05 | [21] |
| N-methylacetamide | CH3CN | 0.2 M NaClO4 | GC | 1.81 | [12] |
| N-acetylethylenediamine | EtOH | 0.1 M LiClO4 | GC | 1.27 | [20] |
| dopamine | EtOH | 0.1 M LiClO4 | GC | 1.22 | [20] |
| N-(5-aminopentyl)biotinamide | EtOH | 0.1 M LiClO4 | GC | 1.22 | [20] |
| diethylamine | CH3CN | 0.1 M [Bu4N][PF6] | GC | 1.10 | [18] |
| dipropylamine | CH3CN | 0.1 M NaClO4 | Pt | 1.00 | [10] |
| dibutylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.11 | [16] |
| THF | 0.1 M LiClO4 | GC | 0.94 | [21] | |
| CH3CN | 0.1 M NaClO4 | Pt | 1.07 | [10] | |
| dibutylamide | THF | 0.1 M LiClO4 | GC | −0.12 | [21] |
| dibenzylamine | CH3CN | 0.1 M NaClO4 | Pt | 1.23 | [10] |
| di-isopropylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.15 | [16] |
| di-isobutylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.11 | [16] |
| di-sec-butylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.16 | [16] |
| CH3CN | 0.1 M NaClO4 | Pt | 1.16 | [10] | |
| dipentylamine | CH3CN | 0.1 M NaClO4 | Pt | 1.11 | [10] |
| bis-2-ethylhexylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.07 | [16] |
| N-methylbutylamine | EtOH | 0.1 M LiClO4 | GC | 1.00 | [20] |
| N-ethylbutylamine | EtOH | 0.1 M LiClO4 | GC | 0.99 | [20] |
| dicyclohexylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.06 | [16] |
| CH3CN | 0.1 M [Bu4N][PF6] | GC | 1.49 | [17] | |
| N,N-dimethylacetamide | CH3CN | 0.2 M NaClO4 | GC | 1.32 | [12] |
| trimethylamine | CH3CN | 0.1 M NaClO4 | Pt | 1.05 | [10] |
| triethylamine | EtOH | 0.1 M LiClO4 | GC | 0.83 | [20] |
| DMF | 0.1 M [Bu4N][BF4] | GC | 0.94 | [16] | |
| CH3CN | 0.1 M [Bu4N][PF6] | GC | 0.88 | [18] | |
| CH3CN | 0.1 M NaClO4 | Pt | 0.95 | [10] | |
| tripropylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 0.95 | [16] |
| CH3CN | 0.1 M NaClO4 | Pt | 0.93 | [10] | |
| tributylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 0.88 | [16] |
| CH3CN | 0.1 M NaClO4 | Pt | 0.78 | [10] | |
| tripentylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 0.91 | [16] |
| CH3CN | 0.1 M NaClO4 | Pt | 0.89 | [10] | |
| tribenzylamine | CH3CN | 0.1 M NaClO4 | Pt | 0.99 | [10] |
| tri-isopropylamine | THF | 0.1 M [Bu4N]ClO4 | GC | 0.76 | [19] |
| tri-isobutylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 0.98 | [16] |
| N,N-dicyclohexylmethylamine | CH3CN | 0.1 M [Bu4N][PF6] | GC | 1.04 | [17] |
| N,N-dimethylcyclohexylamine | CH3CN | 0.1 M [Bu4N][PF6] | GC | 1.18 | [17] |
| N,N-dimethylbutylamine | EtOH | 0.1 M LiClO4 | GC | 0.99 | [20] |
| 4-nitrobenzylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.42 | [16] |
| CH3CN | 0.1 M [Bu4N][BF4] | GC | 1.58 | [16] | |
| 3-nitrobenzylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.51 | [16] |
| CH3CN | 0.1 M [Bu4N][BF4] | GC | 1.78 | [16] | |
| N-methyl-3-nitrobenzylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.25 | [16] |
| CH3CN | 0.1 M [Bu4N][BF4] | GC | 1.33 | [16] | |
| N,N-dimethyl-3-nitrobenzylamine | DMF | 0.1 M [Bu4N][BF4] | GC | 1.01 | [16] |
| CH3CN | 0.1 M [Bu4N][BF4] | GC | 1.07 | [16] | |
| pyrrolidine | CH3CN | 0.1 M [Bu4N][PF6] | GC | 1.16 | [18] |
| pyrrole | CH3CN | 0.5 M NaClO4 | Pt | 1.06 * | [22] |
| pyridine | CH3CN | 0.5 M NaClO4 | Pt | 2.12 * | [22] |
| N,N-dipropylpropionamide | CH3CN | 0.2 M NaClO4 | GC | 1.26 | [12] |
| aniline | CH3CN | 0.5 M NaClO4 | Pt | 0.90 * | [23] |
| p-nitroaniline | CH3CN | 0.5 M NaClO4 | Pt | 1.39 * | [23] |
| p-bromoaniline | CH3CN | 0.5 M NaClO4 | Pt | 0.97 * | [23] |
| p-chloroaniline | CH3CN | 0.5 M NaClO4 | Pt | 0.96 * | [23] |
| p-anisidine | CH3CN | 0.5 M NaClO4 | Pt | 0.62 * | [23] |
| o-anisidine | CH3CN | 0.5 M NaClO4 | Pt | 0.70 * | [23] |
| diphenylamine | CH3CN | 0.1 M NaClO4 | Pt | 0.83 * | [24] |
| triphenylamine | CH3CN | 0.1 M [Et4N]ClO4 | Pt | 0.98 | [25] |
| N,N-dimethylaniline | CH3CN | 0.1 M [Bu4N][PF6] | Pt | 0.76 | [26] |
| N,N-diethylaniline | CH3CN | 0.5 M NaClO4 | Pt | 0.70 * | [23] |
| N,N-diethyl-p-chloroaniline | CH3CN | 0.5 M NaClO4 | Pt | 0.83 * | [23] |
| N,N-dimethyl-p-chloroaniline | CH3CN | 0.5 M NaClO4 | Pt | 0.85 * | [23] |
| ethylphenylamine | CH3CN | 0.5 M NaClO4 | Pt | 0.76 * | [23] |
| di-4-tolylamine | CH3CN | 0.1 M NaClO4 | Pt | 0.70 * | [24] |
| N,N-tetramethylbenzidine | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.43 * | [27] |
| 1-dimethylaminonaphthalene | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.75 * | [27] |
| 2-dimethylaminonaphthalene | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.67 * | [27] |
| azobenzene | CH3CN | 0.5 M NaClO4 | Pt | 1.69 * | [23] |
| 4,4-dichloroazobenzene | CH3CN | 0.5 M NaClO4 | Pt | 1.80 * | [23] |
| 4,4-dimethoxyazobenzene | CH3CN | 0.5 M NaClO4 | Pt | 1.34 * | [23] |
| N,N,N′,N′-tetramethyl-m-phenylenediamine | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.62 * | [28] |
| N,N,N′,N′-tetramethyl-p-phenylenediamine | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.20 * | [28] |
| N,N-dimethyl-m-anisidine | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.79 * | [28] |
| N,N-dimethyl-p-anisidine | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.63 * | [28] |
| 3,4-dimethoxy-N,N-dimethylaniline | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.50 * | [28] |
| 3,5-dimethoxy-N,N-dimethylaniline | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.80 * | [28] |
| N,N,N′,N′-tetramethyl-o-phenylenediamine | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.58 * | [28] |
| N,N-dimethyl-o-anisidine | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.78 * | [28] |
| 2,4-dimethoxy-N,N-dimethylaniline | CH3CN | 0.1 M [Pr4N]ClO4 | Pt | 0.57 * | [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mruthunjaya, A.K.V.; Torriero, A.A.J. Mechanistic Aspects of the Electrochemical Oxidation of Aliphatic Amines and Aniline Derivatives. Molecules 2023, 28, 471. https://doi.org/10.3390/molecules28020471
Mruthunjaya AKV, Torriero AAJ. Mechanistic Aspects of the Electrochemical Oxidation of Aliphatic Amines and Aniline Derivatives. Molecules. 2023; 28(2):471. https://doi.org/10.3390/molecules28020471
Chicago/Turabian StyleMruthunjaya, Ashwin K. V., and Angel A. J. Torriero. 2023. "Mechanistic Aspects of the Electrochemical Oxidation of Aliphatic Amines and Aniline Derivatives" Molecules 28, no. 2: 471. https://doi.org/10.3390/molecules28020471