


Interface-Based Design of High-Affinity Affibody Ligands for the Purification of RBD from Spike Proteins
Abstract
:1. Introduction
2. Results and Discussion
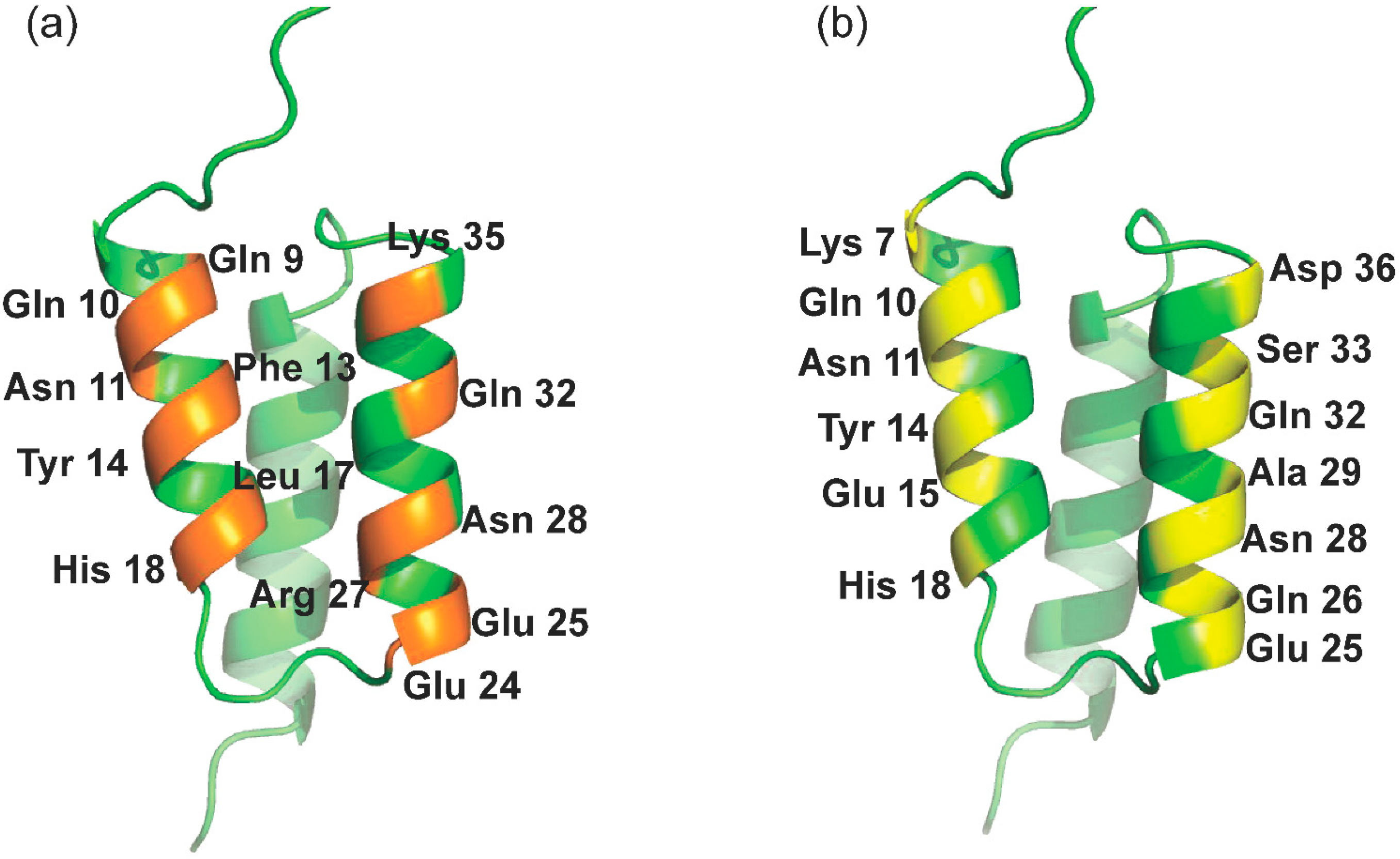
2.1. Library Design and Docking to RBD
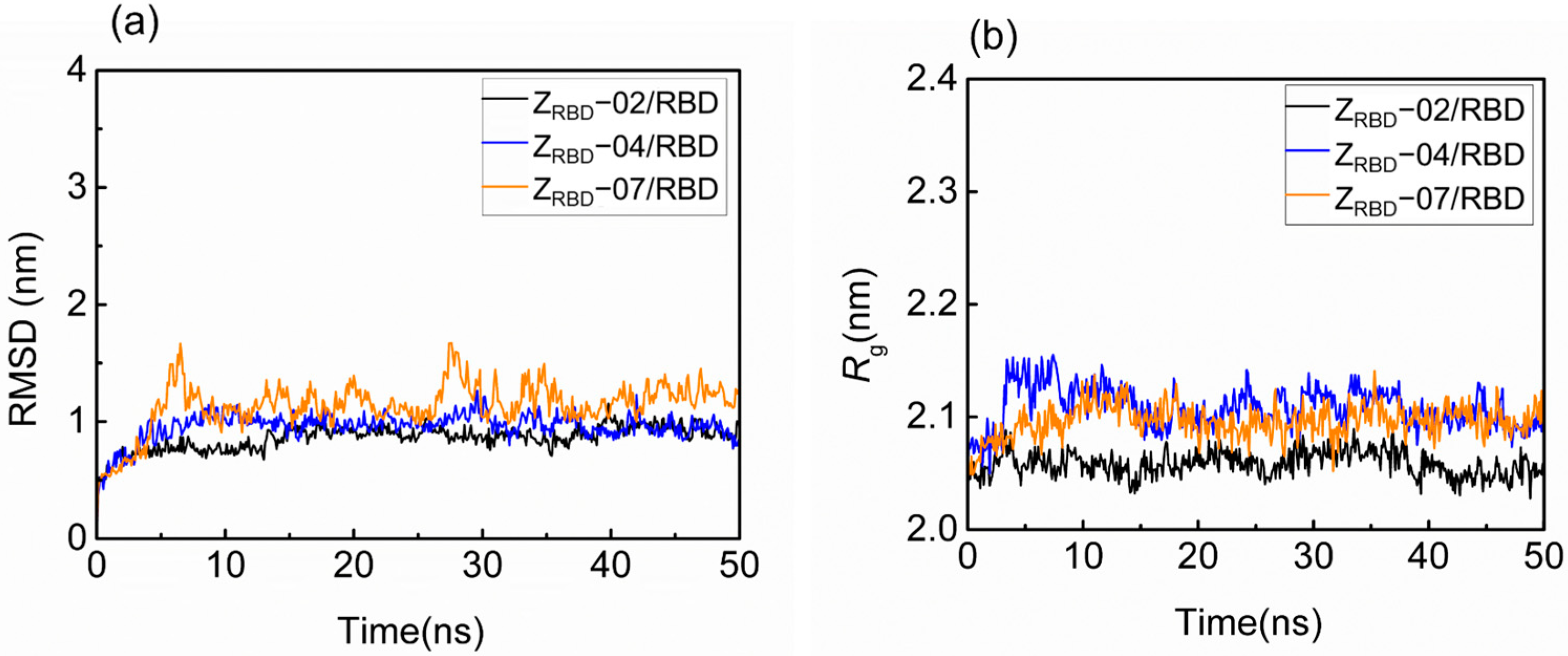
2.2. MD Simulation
2.2.1. Structural Characteristics by MD Simulation
2.2.2. Binding Free Energy Analysis
2.3. ZRBD Characteristics and Binding Affinity
2.3.1. Spectral Characteristics of the ZRBD Affibody
2.3.2. Binding Affinity of the ZRBD Affibody to the RBD
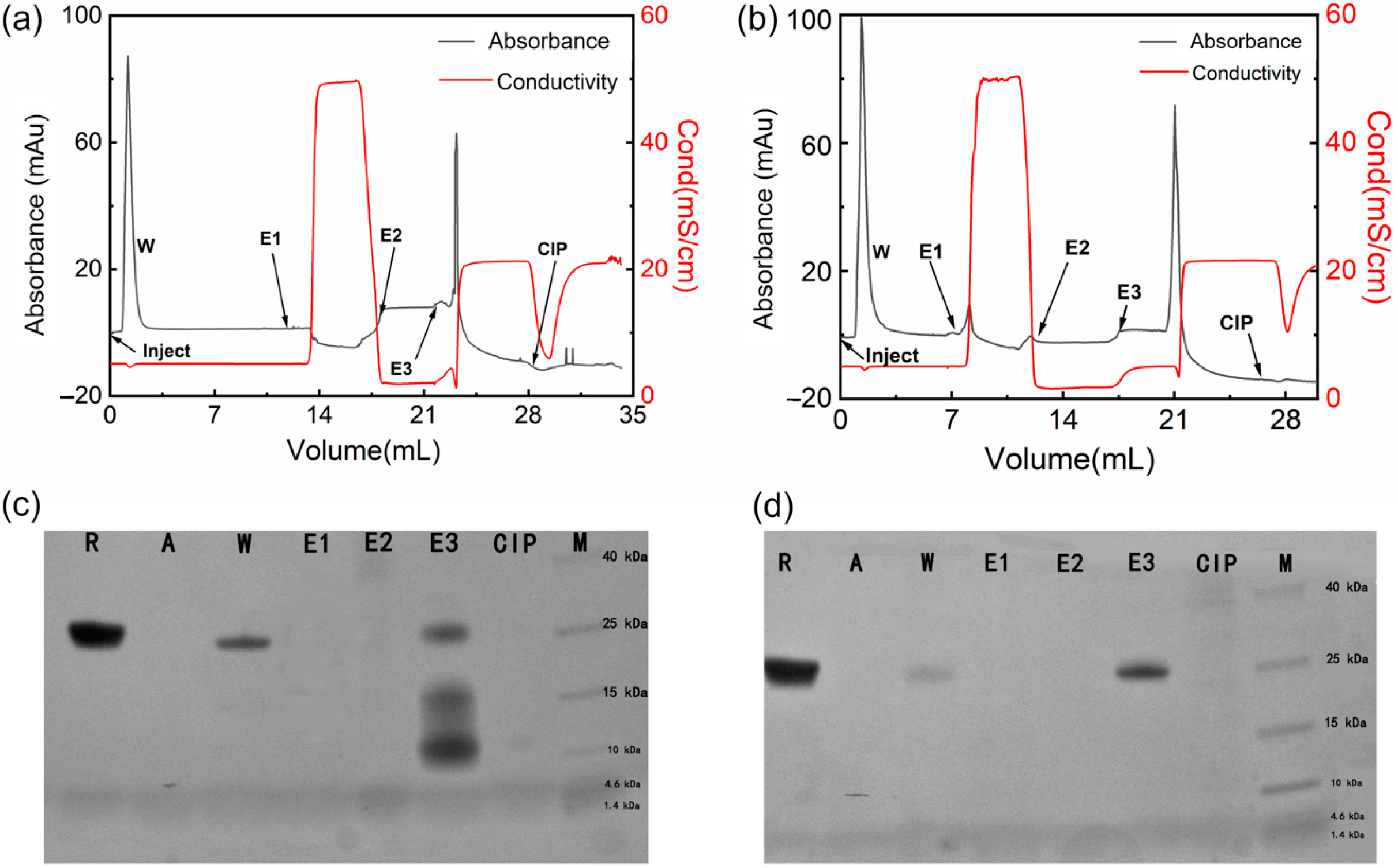
2.4. Chromatographic Performance
3. Materials and Methods
3.1. Materials and Chemicals
3.2. Affibody Modeling
3.3. Docking of ZRBD Affibody to the RBD of Spike Protein
3.4. Molecular Dynamics Simulation
3.5. Spectral Characterization of ZRBD
3.6. Binding Affinity Experiments
3.7. Synthesis of Affibody-Based Gels and Chromatographic Performance
3.8. Sample Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wu, Y.-C.; Chen, C.-S.; Chan, Y.-J. The outbreak of COVID-19: An overview. J. Chin. Med. Assoc. 2020, 83, 217–220. [Google Scholar] [CrossRef]
- Johns Hopkins Coronavirus Resource Center. Coronavirus COVID-19 Global Cases. Available online: https://coronavirus.jhu.edu/map.html (accessed on 30 March 2023).
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.-Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Costa, C.F.S.; Barbosa, A.J.M.; Dias, A.M.G.C.; Roque, A.C.A. Native, engineered and de novo designed ligands targeting the SARS-CoV-2 spike protein. Biotechnol. Adv. 2022, 59, 107986. [Google Scholar] [CrossRef]
- Muratov, E.N.; Amaro, R.; Andrade, C.H.; Brown, N.; Ekins, S.; Fourches, D.; Isayev, O.; Kozakov, D.; Medina-Franco, J.L.; Merz, K.M.; et al. A critical overview of computational approaches employed for COVID-19 drug discovery. Chem. Soc. Rev. 2021, 50, 9121–9151. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.; Franck, C.; Christie, M.; Hawkins, P.M.E.; Patel, K.; Ashhurst, A.S.; Aggarwal, A.; Low, J.K.K.; Siddiquee, R.; Ashley, C.L.; et al. Discovery of Cyclic Peptide Ligands to the SARS-CoV-2 Spike Protein Using mRNA Display. ACS Cent. Sci. 2021, 7, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, W.; Yang, Y.; Zhao, Q.; Yang, C.; Jia, X.; Liu, Y.; Zhou, M.; Zeng, W.; Huang, X.; et al. Developing Next-Generation Protein-Based Vaccines Using High-Affinity Glycan Ligand-Decorated Glyconanoparticles. Adv. Sci. 2023, 10, 202204598. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. SARS-CoV-2 vaccines in development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef]
- Joe, C.C.D.; Chopra, N.; Nestola, P.; Niemann, J.; Douglas, A.D. Rapid-response manufacturing of adenovirus-vectored vaccines. Nat. Biotechnol. 2023, 41, 314–316. [Google Scholar] [CrossRef]
- Zhao, M.; Vandersluis, M.; Stout, J.; Haupts, U.; Sanders, M.; Jacquemart, R. Affinity chromatography for vaccines manufacturing: Finally ready for prime time? Vaccine 2019, 37, 5491–5503. [Google Scholar] [CrossRef]
- Moleirinho, M.G.; Silva, R.J.S.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Current challenges in biotherapeutic particles manufacturing. Expert Opin. Biol. Ther. 2020, 20, 451–465. [Google Scholar] [CrossRef]
- Carvalho, S.B.; Peixoto, C.; Carrondo, M.J.T.; Silva, R.J.S. Downstream processing for influenza vaccines and candidates: An update. Biotechnol. Bioeng. 2021, 118, 2845–2869. [Google Scholar] [CrossRef]
- Wang, Q.; Lock, M.; Prongay, A.J.; Alvira, M.R.; Petkov, B.; Wilson, J.M. Identification of an adeno-associated virus binding epitope for AVB sepharose affinity resin. Mol. Ther. Methods Clin. Dev. 2015, 2, 15040. [Google Scholar] [CrossRef]
- Van Lieshout, L.P.; Stegelmeier, A.A.; Rindler, T.N.; Lawder, J.J.; Sorensen, D.L.; Frost, K.L.; Booth, S.A.; Bridges, J.P.; Wootton, S.K. Engineered AAV8 capsid acquires heparin and AVB sepharose binding capacity but has altered in vivo transduction efficiency. Gene Ther. 2023, 30, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; De Vlieger, D.; Corbett, K.S.; Torres, G.M.; Wang, N.; Van Breedam, W.; Roose, K.; van Schie, L.; Hoffmann, M.; Pöhlmann, S.; et al. Structural Basis for Potent Neutralization of Betacoronaviruses by Single-Domain Camelid Antibodies. Cell 2020, 181, 1004–1015.e15. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Hanke, L.; Perez, L.V.; Sheward, D.J.; Das, H.; Schulte, T.; Moliner-Morro, A.; Corcoran, M.; Achour, A.; Hedestam, G.B.K.; Haellberg, B.M.; et al. An alpaca nanobody neutralizes SARS-CoV-2 by blocking receptor interaction. Nat. Commun. 2020, 11, 4420. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Chen, F.-J.; Li, K.; Reja, R.M.; Haeffner, F.; Gao, J. Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display. J. Am. Chem. Soc. 2022, 144, 15885–15893. [Google Scholar] [CrossRef]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 27, 846–854. [Google Scholar] [CrossRef]
- Yang, F.; Liu, L.; Neuenschwander, P.F.; Idell, S.; Vankayalapati, R.; Jain, K.G.; Du, K.; Ji, H.; Yi, G. Phage Display-Derived Peptide for the Specific Binding of SARS-CoV-2. ACS Omega 2022, 7, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, A.; Glasgow, J.; Limonta, D.; Solomon, P.; Lui, I.; Zhang, Y.; Nix, M.A.; Rettko, N.J.; Zha, S.; Yamin, R.; et al. Engineered ACE2 receptor traps potently neutralize SARS-CoV-2. Proc. Natl. Acad. Sci. USA. 2020, 117, 28046–28055. [Google Scholar] [CrossRef]
- Szardenings, M. Phage display of random peptide libraries: Applications, limits, and potential. J. Recept. Signal Transduct. 2003, 23, 307–349. [Google Scholar] [CrossRef]
- Rodi, D.J.; Makowski, L. Phage-display technology—Finding a needle in a vast molecular haystack. Curr. Opin. Biotechnol. 1999, 10, 87–93. [Google Scholar] [CrossRef]
- Marshall, S.A.; Lazar, G.A.; Chirino, A.J.; Desjarlais, J.R. Rational design and engineering of therapeutic proteins. Drug Discov. Today 2003, 8, 212–221. [Google Scholar] [CrossRef]
- Zhao, W.-W.; Liu, F.-F.; Shi, Q.-H.; Sun, Y. Octapeptide-based affinity chromatography of human immunoglobulin G: Comparisons of three different ligands. J. Chromatogr. A 2014, 1359, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.-F.; Wang, T.; Dong, X.-Y.; Sun, Y. Rational design of affinity peptide ligand by flexible docking simulation. J. Chromatogr. A 2007, 1146, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.-M.; Lin, D.-Q.; Yao, S.-J. Review on biomimetic affinity chromatography with short peptide ligands and its application to protein purification. J. Chromatogr. A 2018, 1571, 1–15. [Google Scholar] [CrossRef]
- Orlova, A.; Magnusson, M.; Eriksson, T.L.; Nilsson, M.; Larsson, B.; Höidén-Guthenberg, I.; Widström, C.; Carlsson, J.; Tolmachev, V.; Ståhl, S.; et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res 2006, 66, 4339–4348. [Google Scholar] [CrossRef]
- Chowdhury, S.M.; Talukder, S.A.; Khan, A.M.; Afrin, N.; Ali, M.A.; Islam, R.; Parves, R.; Al Mamun, A.; Abu Sufian, M.; Hossain, M.N.; et al. Antiviral Peptides as Promising Therapeutics against SARS-CoV-2. J. Phys. Chem. B 2020, 124, 9785–9792. [Google Scholar] [CrossRef]
- Wahlberg, E.; Lendel, C.; Helgstrand, M.; Allard, P.; Dincbas-Renqvist, V.; Hedqvist, A.; Berglund, H.; Nygren, P.A.; Hard, T. An affibody in complex with a target protein: Structure and coupled folding. Proc. Natl. Acad. Sci. USA 2003, 100, 3185–3190. [Google Scholar] [CrossRef]
- Lendel, C.; Dogan, J.; Hard, T. Structural basis for molecular recognition in an Affibody: Affibody complex. J. Mol. Biol. 2006, 359, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Nord, K.; Gunneriusson, E.; Uhlen, M.; Nygren, P.A. Ligands selected from combinatorial libraries of protein A for use in affinity capture of apolipoprotein A-1(M) and Taq DNA polymerase. J. Biotechnol. 2000, 80, 45–54. [Google Scholar] [CrossRef]
- Malm, M.; Bass, T.; Gudmundsdotter, L.; Lord, M.; Frejd, F.Y.; Stahl, S.; Lofblom, J. Engineering of a bispecific affibody molecule towards HER2 and HER3 by addition of an albumin-binding domain allows for affinity purification and in vivo half-life extension. Biotechnol. J. 2014, 9, 1215–1222. [Google Scholar] [CrossRef]
- Antaris, A.L.; Chen, H.; Cheng, K.; Sun, Y.; Hong, G.; Qu, C.; Diao, S.; Deng, Z.; Hu, X.; Zhang, B.; et al. A small-molecule dye for NIR-II imaging. Nat. Mater. 2016, 15, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Stahl, S.; Graslund, T.; Karlstrom, A.E.; Frejd, F.Y.; Nygren, P.-A.; Lofblom, J. Affibody Molecules in Biotechnological and Medical Applications. Trends Biotechnol. 2017, 35, 691–712. [Google Scholar] [CrossRef] [PubMed]
- Repligen and Navigo Proteins Announce the Launch of an Affinity Resin for the Purification of COVID-19 Vaccines. Available online: https://www.navigo-proteins.com/repligen-and-navigo-proteins-announce-the-launch-of-an-affinity-resin-for-the-purification-of-covid-19-vaccines/ (accessed on 8 July 2023).
- Nilsson, B.; Moks, T.; Jansson, B.; Abrahmsen, L.; Elmblad, A.; Holmgren, E.; Henrichson, C.; Jones, T.A.; Uhlen, M. A synthetic IgG-binding domain based on staphylococcal protein A. Protein Eng. 1987, 1, 107–113. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.J.; Van Dijk, A.D.; Krzeminski, M.; van Dijk, M.; Thureau, A.; Hsu, V.; Wassenaar, T.; Bonvin, A.M. HADDOCK versus HADDOCK: New features and performance of HADDOCK2.0 on the CAPRI targets. Proteins Struct. Funct. Bioinform. 2007, 69, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef]
- Baumketner, A.; Shea, J.-E. The structure of the Alzheimer amyloid β 10-35 peptide probed through replica-exchange molecular dynamics simulations in explicit solvent. J. Mol. Biol. 2007, 366, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Consortium, O.S.D.D.; Lynn, A. g_mmpbsa-A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Wang, Z.; Shen, Y.; Shi, Q.-H.; Sun, Y. Insights into the molecular structure of immobilized protein A ligands on dextran-coated nanoparticles: Comprehensive spectroscopic investigation. Biochem. Eng. J. 2019, 146, 20–30. [Google Scholar] [CrossRef]
- Wang, Q.; Dou, X.; Chen, X.; Zhao, Z.; Wang, S.; Wang, Y.; Sui, K.; Tan, Y.; Gong, Y.; Zhang, Y. Reevaluating protein photoluminescence: Remarkable visible luminescence upon concentration and insight into the emission mechanism. Angew. Chem. 2019, 131, 12797–12803. [Google Scholar] [CrossRef]
- Pomplun, S.; Jbara, M.; Quartararo, A.J.; Zhang, G.; Brown, J.S.; Lee, Y.-C.; Ye, X.; Hanna, S.; Pentelute, B.L. De novo discovery of high-affinity peptide binders for the SARS-CoV-2 spike protein. ACS Cent. Sci. 2020, 7, 156–163. [Google Scholar] [CrossRef]
- Dutta, A.; Utturkar, A.; Tchelet, R.; Crespo, N.V.; Mueller, L.; Jacquemart, R. Affinity purification of SARS-CoV-2 spike protein receptor binding domain produced in a C1 fungal expression system. In Proceedings of the VACCINE TECHNOLOGY III, Sitges, Spain, 12 June 2022. [Google Scholar]
- Fu, D.; Zhang, G.; Wang, Y.; Zhang, Z.; Hu, H.; Shen, S.; Wu, J.; Li, B.; Li, X.; Fang, Y. Structural basis for SARS-CoV-2 neutralizing antibodies with novel binding epitopes. PLoS Biol. 2021, 19, e3001209. [Google Scholar] [CrossRef]
- Nord, K.; Gunneriusson, E.; Ringdahl, J.; Stahl, S.; Uhlen, M.; Nygren, P.A. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat. Biotechnol. 1997, 15, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.D.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Sun, Y. Biomimetic Design of Platelet Adhesion Inhibitors to Block Integrin alpha 2 beta 1-Collagen Interactions: I. Construction of an Affinity Binding Model. Langmuir 2014, 30, 4725–4733. [Google Scholar] [CrossRef]
- Shen, Y.; Chu, X.; Shi, Q. Unraveling structure and performance of protein a ligands at liquid-solid interfaces: A multi-techniques analysis. Chin. J. Chem. Eng. 2023, 54, 232–239. [Google Scholar] [CrossRef]
- Ferraz, N.; Leverrier, J.; Batista-Viera, F.; Manta, C. Thiopropyl-Agarose as a Solid Phase Reducing Agent for Chemical Modification of IgG and F(ab’)2. Biotechnol. Prog. 2008, 24, 1154–1159. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Affibody Modules |
|---|---|
| ZRBD-01 | K4Q |
| ZRBD-02 | K4Q, E24Q, Q26T, A29D, F30K, Q32H, S33E |
| ZRBD-03 | K4Q, K7H, Q10T, N11Y, Y14Q, E15D, I16K |
| ZRBD-04 | K4G |
| ZRBD-05 | K4G, K7D |
| ZRBD-06 | K4G, K7D, H18F |
| ZRBD-07 | K4Q, K7D |
| ZRBD-08 | K4Q, Q10E |
| Complex | HADDOCK Score | Van der Waals Energy (kJ/mol) | Electrostatic Energy (kJ/mol) | Desolvation Energy (kJ/mol) | Restraints Violation Energy (kJ/mol) | Buried Surface Area (Å2) |
|---|---|---|---|---|---|---|
| ZRBD/RBD | −108.6 ± 2.6–97.8 ± 2.4 | −66.8 ± 7.0 | −204.4 ± 43.9 | −14.3 ± 4.3 | 45.1 ± 38.6 | 1741.9 ± 49.9 |
| ZRBD-01/RBD | −97.8 ± 2.4 | −51.3 ± 5.2 | −161.9 ± 30.6 | −17.7 ± 1.5 | 36.1 ± 20.6 | 1567.6 ± 36.2 |
| ZRBD-02/RBD | −137.6 ± 5.2 | −80.6 ± 8.2 | −243.4 ± 27.8 | −23.7 ± 1.4 | 153.8 ± 14.2 | 2094.8 ± 76.4 |
| ZRBD-03/RBD | −74.1 ± 5.8 | −75.9 ± 2.1 | −276.2 ± 11.3 | −22.2 ± 4.0 | 79.24 ± 9.15 | 2093.6 ± 49.0 |
| ZRBD-04/RBD | −132.7 ± 1.9 | −71.6 ± 7.0 | −290.0 ± 25.8 | −19.1 ± 1.5 | 160.6 ± 17.6 | 2137.7 ± 40.7 |
| ZRBD-05/RBD | −117.7 ± 3.4 | −60.9 ± 5.8 | −187.2 ± 41.8 | −22.9 ± 3.0 | 34.3 ± 20.9 | 1679.5 ± 37.3 |
| ZRBD-06/RBD | −114.1 ± 3.5 | −65.1 ± 3.6 | −109.8 ± 3.9 | −31.1 ± 2.8 | 41.2 ± 32.0 | 1609.3 ± 50.8 |
| ZRBD-07/RBD | −125.3 ± 7.5 | −65.6 ± 6.7 | −219.8 ± 11.1 | −20.1 ± 1.3 | 43.9 ± 12.4 | 1756.0 ± 91.0 |
| ZRBD-08/RBD | −109.5 ± 13.2 | −57.2 ± 9.3 | −184.8 ± 55.9 | −21.4 ± 6.9 | 61.0 ± 36.8 | 1805.6 ± 152.2 |
| ACE2/RBD | −117.3 ± 3.6 | −52.6 ± 4.0 | −261.2 ± 42.2 | −15.9 ± 5.1 | 34.1 ± 20.8 | 1840.3 ± 39.6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, S.; Shi, Q. Interface-Based Design of High-Affinity Affibody Ligands for the Purification of RBD from Spike Proteins. Molecules 2023, 28, 6358. https://doi.org/10.3390/molecules28176358
Song S, Shi Q. Interface-Based Design of High-Affinity Affibody Ligands for the Purification of RBD from Spike Proteins. Molecules. 2023; 28(17):6358. https://doi.org/10.3390/molecules28176358
Chicago/Turabian StyleSong, Siyuan, and Qinghong Shi. 2023. "Interface-Based Design of High-Affinity Affibody Ligands for the Purification of RBD from Spike Proteins" Molecules 28, no. 17: 6358. https://doi.org/10.3390/molecules28176358