
Effects of Methyl Substitution and Leaving Group on E2/SN2 Competition for Reactions of F− with RY (R = CH3, C2H5, iC3H7, tC4H9; Y = Cl, I)

Abstract
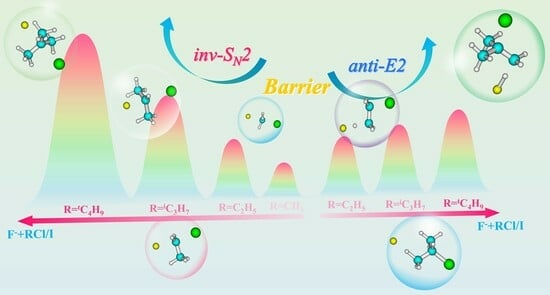
:
1. Introduction
2. Results and Discussion
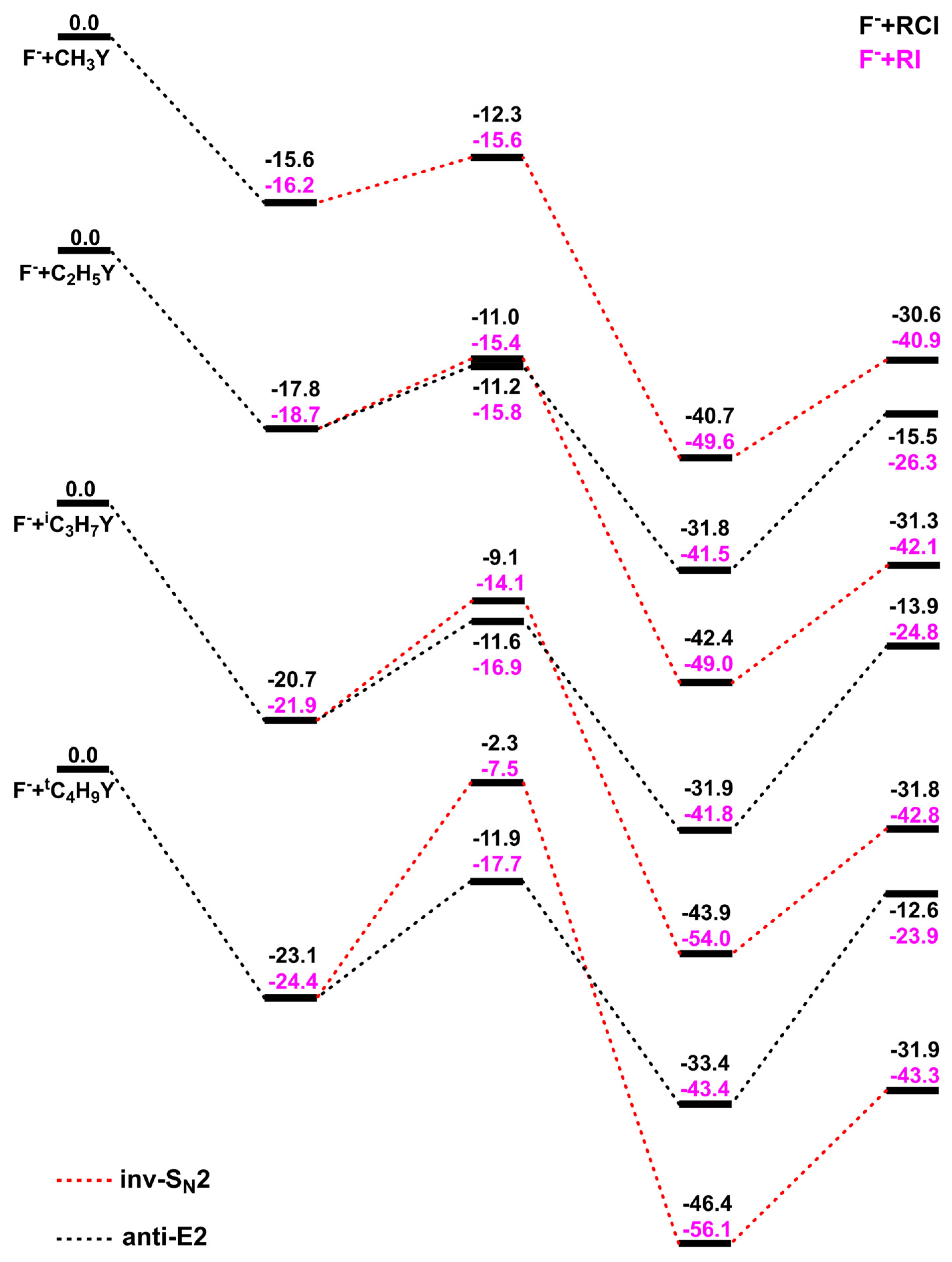
2.1. Potential Energy Surfaces of F− + RY Reactions
2.2. Effects of α-Methyl Substitution
2.3. Effects of Leaving Group
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Villano, S.M.; Eyet, N.; Lineberger, W.C.; Bierbaum, V.M. Reactions of α-Nucleophiles with Alkyl Chlorides: Competition between SN2 and E2 Mechanisms and the Gas-Phase α-Effect. J. Am. Chem. Soc. 2009, 131, 8227–8233. [Google Scholar] [CrossRef] [PubMed]
- Garver, J.M.; Fang, Y.R.; Eyet, N.; Villano, S.M.; Bierbaum, V.M.; Westaway, K.C. A Direct Comparison of Reactivity and Mechanism in the Gas Phase and in Solution. J. Am. Chem. Soc. 2010, 132, 3808–3814. [Google Scholar] [CrossRef] [PubMed]
- Carrascosa, E.; Meyer, J.; Michaelsen, T.; Stei, M.; Wester, R. Conservation of direct dynamics in sterically hindered SN2/E2 reactions. Chem. Sci. 2018, 9, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Gronert, S. Theoretical Studies of Elimination Reactions. 1. Reactions of F− and PH2− with CH3CH2Cl. Competition between SN2 and E2 Mechanisms for First- and Second-Row Nucleophiles. J. Am. Chem. Soc. 1991, 113, 6041–6048. [Google Scholar] [CrossRef]
- Haib, J.; Stahl, D. Competition Between Substitution (SN2), Elimination (E2) and Addition Elimination (AE) Reactions in the Gas Phase. Org. Mass. Spectrom. 1992, 27, 377–382. [Google Scholar] [CrossRef]
- Hu, W.P.; Truhlar, D.G. Factors Affecting Competitive Ion-Molecule Reactions: ClO− + C2H5Cl and C2D5Cl via E2 and SN2 Channels. J. Am. Chem. Soc. 1996, 118, 860–869. [Google Scholar] [CrossRef]
- Glad, S.S.; Jensen, F. Kinetic Isotope Effects and Transition State Geometries. A Theoretical Investigation of E2 Model Systems. J. Org. Chem. 1997, 62, 253–260. [Google Scholar] [CrossRef]
- Chung, D.S.; Kim, C.K.; Lee, I. Theoretical Studies of Competitive Gas-Phase SN2 and E2 Reactions of NCCH2CH2Cl with OH− and SH−. J. Phys. Chem. A 1997, 101, 9097–9104. [Google Scholar] [CrossRef]
- Bickelhaupt, F.M. Understanding Reactivity with Kohn-Sham Molecular Orbital Theory: E2-SN2 Mechanistic Spectrum and Other Concepts. J. Comput. Chem. 1999, 20, 114–128. [Google Scholar] [CrossRef]
- Mugnai, M.; Cardini, G.; Schettino, V. Substitution and Elimination Reaction of F− with C2H5Cl: An ab Initio Molecular Dynamics Study. J. Phys. Chem. A 2003, 107, 2540–2547. [Google Scholar] [CrossRef]
- Vayner, G.; Houk, K.N.; Jorgensen, W.L.; Brauman, J.I. Steric Retardation of SN2 Reactions in the Gas Phase and Solution. J. Am. Chem. Soc. 2004, 126, 9054–9058. [Google Scholar] [CrossRef]
- Ochran, R.A.; Uggerud, E. SN2 reactions with allylic substrates—Trends in reactivity. Int. J. Mass. Spectrom. 2007, 265, 169–175. [Google Scholar] [CrossRef]
- Bento, A.P.; Solà, M.; Bickelhaupt, F.M. E2 and SN2 Reactions of X− + CH3CH2X(X = F, Cl); an ab Initio and DFT Benchmark Study. J. Chem. Theory Comput. 2008, 4, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.P.; Sun, X.M.; Wei, X.G.; Ren, Y.; Wong, N.B.; Li, W.K. Exploring the Reactivity Trends in the E2 and SN2 Reactions of X− + CH3CH2Cl (X = F, Cl, Br, HO, HS, HSe, NH2, PH2, AsH2,CH3, SiH3, and GeH3). J. Chem. Theory Comput. 2009, 5, 1597–1606. [Google Scholar] [CrossRef]
- Wolters, L.P.; Ren, Y.; Bickelhaup, F.M. Understanding E2 versus SN2 Competition under Acidic and Basic Conditions. ChemistryOpen 2014, 3, 29–36. [Google Scholar] [CrossRef]
- Tajti, V.; Czakó, G. Benchmark ab Initio Characterization of the Complex Potential Energy Surface of the F− + CH3CH2Cl Reaction. J. Phys. Chem. A 2017, 121, 2847–2854. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, J.X.; Xie, J.; Ma, X.Y.; Zhang, L.Y.; Zhao, C.Y.; Hase, W.L. Competing E2 and SN2 Mechanisms for the F− + CH3CH2I Reaction. J. Phys. Chem. A 2017, 121, 1078–1085. [Google Scholar] [CrossRef]
- Hamlin, T.A.; Swart, M.; Bickelhaupt, F.M. Nucleophilic Substitution (SN2): Dependence on Nucleophile, Leaving Group, Central Atom, Substituents, and Solvent. Chemphyschem 2018, 19, 1315–1330. [Google Scholar] [CrossRef] [PubMed]
- Satpathy, L.; Sahu, P.K.; Behera, P.K.; Mishra, B.K. Solvent Effect on the Potential Energy Surfaces of the F− + CH3CH2Br Reaction. J. Phys. Chem. A 2018, 122, 5861–5869. [Google Scholar] [CrossRef]
- Garver, J.M.; Eyet, N.; Villano, S.M.; Yang, Z.; Bierbaum, V.M. Mechanistic investigation of SN2 dominated gas phase alkyl iodide reactions. Int. J. Mass. Spectrom. 2011, 301, 151–158. [Google Scholar] [CrossRef]
- Vermeeren, P.; Hansen, T.; Jansen, P.; Swart, M.; Hamlin, T.A.; Bickelhaupt, F.M. A Unified Framework for Understanding Nucleophilicity and Protophilicity in the SN2/E2 Competition. Chem. Eur. J. 2020, 26, 15538–15548. [Google Scholar] [CrossRef] [PubMed]
- Vermeeren, P.; Hansen, T.; Grasser, M.; Silva, D.R.; Hamlin, T.A.; Bickelhaupt, F.M. SN2 versus E2 Competition of F− and PH2− Revisited. J. Org. Chem. 2020, 85, 14087–14093. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.; Vermeeren, P.; Bickelhaupt, F.M.; Hamlin, T.A. Origin of the α-Effect in SN2 Reactions. Angew. Chem. Int. Ed. 2021, 60, 20840–20848. [Google Scholar] [CrossRef] [PubMed]
- Stuyver, T.; Shaik, S. Resolving Entangled Reactivity Modes through External Electric Fields and Substitution: Application to E2/SN2 Reactions. J. Org. Chem. 2021, 86, 9030–9039. [Google Scholar] [CrossRef] [PubMed]
- Tasi, D.A.; Tokaji, C.; Czako, G. A benchmark ab initio study of the complex potential energy surfaces of the OH− + CH3CH2Y [Y = F, Cl, Br, I] reactions. Phys. Chem. Chem. Phys. 2021, 23, 13526–13534. [Google Scholar] [CrossRef]
- Hansen, T.; Roozee, J.C.; Bickelhaupt, F.M.; Hamlin, T.A. How Solvation Influences the SN2 versus E2 Competition. J. Org. Chem. 2022, 87, 1805–1813. [Google Scholar] [CrossRef]
- Li, Y.; Li, C.; Gao, D.; Wang, D. Atomic-Level Mechanism, Solvent Effect, and Potential of the Mean Force of the F− + CH3CH2Cl SN2 Reaction in Aqueous Solution. J. Phys. Chem. A 2022, 126, 5527–5533. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, S.; Xie, J. Investigating the competing E2 and SN2 mechanisms for the microsolvated HO− (H2O)n=0–4 + CH3CH2X (X = Cl, Br, I) reactions. Phys. Chem. Chem. Phys. 2022, 24, 12993–13005. [Google Scholar] [CrossRef]
- Zhao, S.; Fu, G.; Zhen, W.; Yang, L.; Sun, J.; Zhang, J. Reaction mechanism conversion induced by the contest of nucleophile and leaving group. Phys. Chem. Chem. Phys. 2022, 24, 24146–24154. [Google Scholar] [CrossRef]
- Tajti, V.; Czako, G. Vibrational mode-specific dynamics of the F− + CH3CH2Cl multi-channel reaction. Phys. Chem. Chem. Phys. 2022, 24, 8166–8181. [Google Scholar] [CrossRef]
- Meyer, J.; Tajti, V.; Carrascosa, E.; Gyori, T.; Stei, M.; Michaelsen, T.; Bastian, B.; Czako, G.; Wester, R. Atomistic dynamics of elimination and nucleophilic substitution disentangled for the F− + CH3CH2Cl reaction. Nat. Chem. 2021, 13, 977–981. [Google Scholar] [CrossRef]
- Nettey, S.; Swift, C.A.; Joviliano, R.; Noin, D.O.; Gronert, S. The impact of substituents on the transition states of SN2 and E2 reactions in aliphatic and vinylic systems: Remarkably facile vinylic eliminations. J. Am. Chem. Soc. 2012, 134, 9303–9310. [Google Scholar] [CrossRef] [PubMed]
- Martinez, H.; Rebeyrol, A.; Nelms, T.B.; Dolbier, W.R. Impact of fluorine substituents on the rates of nucleophilic aliphatic substitution and β-elimination. J. Fluor. Chem. 2012, 135, 167–175. [Google Scholar] [CrossRef]
- Conner, K.M.; Gronert, S. Impact of alkyl substituents on the gas-phase competition between substitution and elimination. J. Org. Chem. 2013, 78, 8606–8613. [Google Scholar] [CrossRef] [PubMed]
- Rablen, P.R.; McLarney, B.D.; Karlow, B.J.; Schneider, J.E. How alkyl halide structure affects E2 and SN2 reaction barriers: E2 reactions are as sensitive as SN2 reactions. J. Org. Chem. 2014, 79, 867–879. [Google Scholar] [CrossRef]
- DePuy, C.H.; Gronert, S.; Mullin, A.; Bierbaum, V.M. Gas-Phase SN2 and E2 Reactions of Alkyl Halides. J. Am. Chem. Soc. 1990, 112, 8650–8655. [Google Scholar] [CrossRef]
- Gronert, S.; DePuy, C.H.; Bierbaum, V.M. Deuterium Isotope Effects in Gas-Phase Reactions of Alkyl Halides: Distinguishing E2 and SN2 Pathways. J. Am. Chem. Soc. 1991, 113, 4009–4010. [Google Scholar] [CrossRef]
- Gronert, S. Theoretical Studies of Elimination Reactions. 3. Gas-Phase Reactions of F− with (CH3)2CHCl and CH3CH2CH2Cl. The Effect of Methyl Substituents. J. Am. Chem. Soc. 1993, 115, 652–659. [Google Scholar] [CrossRef]
- Gallegos, M.; Costales, A.; Pendás, Á.M. Does Steric Hindrance Actually Govern the Competition between Bimolecular Substitution and Elimination Reactions? J. Phys. Chem. A 2022, 126, 1871–1880. [Google Scholar] [CrossRef]
- Gallegos, M.; Costales, A.; Pendás, Á.M. A real space picture of the role of steric effects in SN2 reactions. J. Comput. Chem. 2022, 43, 785–795. [Google Scholar] [CrossRef]
- Carrascosa, E.; Meyer, J.; Zhang, J.; Stei, M.; Michaelsen, T.; Hase, W.L.; Yang, L.; Wester, R. Imaging dynamic fingerprints of competing E2 and SN2 reactions. Nat. Comun. 2017, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Szabo, I.; Czako, G. Revealing a double-inversion mechanism for the F− + CH3Cl SN2 reaction. Nat. Commun. 2015, 6, 5972. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Carrascosa, E.; Michaelsen, T.; Bastian, B.; Li, A.; Guo, H.; Wester, R. Unexpected Indirect Dynamics in Base-Induced Elimination. J. Am. Chem. Soc. 2019, 141, 20300–20308. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hase, W.L. Electronic Structure Theory Study of the F− + CH3I → FCH3 + I− Potential Energy Surface. J. Phys. Chem. A 2010, 114, 9635–9643. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Császár, A.G.; Czakó, G. Dynamics of the F− + CH3Cl → Cl− + CH3F SN2 reaction on a chemically accurate potential energy surface. Chem. Sci. 2013, 4, 4362. [Google Scholar] [CrossRef]
- Stei, M.; Carrascosa, E.; Kainz, M.A.; Kelkar, A.H.; Meyer, J.; Szabo, I.; Czako, G.; Wester, R. Influence of the leaving group on the dynamics of a gas-phase SN2 reaction. Nat. Chem. 2016, 8, 151–156. [Google Scholar] [CrossRef]
- Olasz, B.; Szabo, I.; Czako, G. High-level ab initio potential energy surface and dynamics of the F− + CH3I SN2 and proton-transfer reactions. Chem. Sci. 2017, 8, 3164–3170. [Google Scholar] [CrossRef]
- Szabo, I.; Czako, G. Benchmark ab Initio Characterization of the Complex Potential Energy Surface of the Cl− + CH3I Reaction. J. Phys. Chem. A 2017, 121, 5748–5757. [Google Scholar] [CrossRef]
- Gyori, T.; Olasz, B.; Paragi, G.; Czako, G. Effects of the Level of Electronic Structure Theory on the Dynamics of the F− + CH3I Reaction. J. Phys. Chem. A 2018, 122, 3353–3364. [Google Scholar] [CrossRef]
- Tasi, D.A.; Fabian, Z.; Czako, G. Benchmark ab Initio Characterization of the Inversion and Retention Pathways of the OH− + CH3Y [Y = F, Cl, Br, I] SN2 Reactions. J. Phys. Chem. A 2018, 122, 5773–5780. [Google Scholar] [CrossRef]
- Tasi, D.A.; Fabian, Z.; Czako, G. Rethinking the X− + CH3Y [X = OH, SH, CN, NH2, PH2; Y = F, Cl, Br, I] SN2 reactions. Phys. Chem. Chem. Phys. 2019, 21, 7924–7931. [Google Scholar] [CrossRef] [PubMed]
- Kerekes, Z.; Tasi, D.A.; Czako, G. SN2 Reactions with an Ambident Nucleophile: A Benchmark Ab Initio Study of the CN− + CH3Y [Y = F, Cl, Br, and I] Systems. J. Phys. Chem. A 2022, 126, 889–900. [Google Scholar] [CrossRef] [PubMed]
- Vermeeren, P.; Hamlin, T.A.; Bickelhaupt, F.M. Chemical reactivity from an activation strain perspective. Chem. Commun. 2021, 57, 5880–5896. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Fu, G.; Zhen, W.; Wang, H.; Liu, M.; Yang, L.; Zhang, J. Nucleophile Effects on the E2/SN2 Competition for the X− + CH3CH2Cl Reactions: A Theoretical Study. J. Phys. Chem. A 2023, 127, 3381–3389. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Kim, C.K.; Li, H.G.; Lee, B.-S.; Kim, C.K.; Lee, H.W.; Lee, I. Gas-Phase Identity Nucleophilic Substitution Reactions of Cyclopropenyl Halides. J. Org. Chem. 2002, 67, 1953–1960. [Google Scholar] [CrossRef]
- Ren, Y.; Yamataka, H. The α-Effect in Gas-Phase SN2 Reactions: Existence and the Origin of the Effect. J. Org. Chem. 2007, 72, 5660–5667. [Google Scholar] [CrossRef]
- Chen, Y.; Chang, K.H.; Meng, F.Y.; Tseng, S.M.; Chou, P.T. Broadening the Horizon of the Bell–Evans–Polanyi Principle towards Optically Triggered Structure Planarization. Angew. Chem. Int. Ed. 2021, 60, 7205–7212. [Google Scholar] [CrossRef]
- Head-Gordon, M.; Pople, J.A. MP2 Eenergy Evaluation By Direct Methods. Chem. Phys. Lett. 1988, 153, 503–506. [Google Scholar] [CrossRef]
- Frisch, M.J.; Head-Gordon, M.; Pople, J.A. Semi-Direct Algorithms for the MP2 Energy and Gradient. Chem. Phys. Lett. 1990, 166, 281–289. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Wadt, W.R.; Hay, P.J. Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 1985, 82, 284–298. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L.; Zhao, Y.; Zhang, J.; Feng, D.; Sun, S. Effects of Water Molecule on CO Oxidation by OH: Reaction Pathways, Kinetic Barriers, and Rate Constants. J. Phys. Chem. A 2017, 121, 4868–4880. [Google Scholar] [CrossRef]
- Szucs, T.; Czako, G. Benchmark ab initio potential energy surface mapping of the F− + CH3NH2 reaction. Phys. Chem. Chem. Phys. 2022, 24, 20249. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Scalmani, G.; Barone, V.; Mennucci, B.; et al. Gaussian 09; Revision A.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cl | I | ||||||
|---|---|---|---|---|---|---|---|
| R | Species | MP2 | CCSD(T) a | Exptl c | MP2 | CCSD(T) b | Exptl c |
| CH3 | 1dRCH | 16.9 | 17.6 | −18.9 | −20.3 | ||
| 1TSRC | −15.1 | −15.8 | −15.8 | −17.6 | |||
| 1bRC | −15.6 | −16.1 | −16.2 | −18.3 | |||
| 1bTS | −12.3 | −12.8 | −15.6 | −18.1 | |||
| 1dTS | 32.1 | 30.7 | 22.8 | 18.3 | |||
| 1dPC | −40.7 | −41.6 | −49.6 | −55.3 | |||
| 1bPC | −40.7 | −41.6 | −49.6 | −55.3 | |||
| 1P2 | −30.6 | −31.8 | −31.1 | −40.9 | −48.0 | −42.3 | |
| C2H5 | 2c/2dRCH | −17.8 | −18.0 | −18.7 | −19.6 | ||
| 2a/2bRC | −17.5 | −17.7 | −19.4 | −20.1 | |||
| 2aTS | −11.2 | −11.1 | −15.8 | −16.0 | |||
| 2bTS | −11 | −11.3 | −15.4 | −16.9 | |||
| 2cTS | −0.1 | −6.8 | −4.2 | −4.9 | |||
| 2dTS | 31.1 | 30.0 | 21.5 | 19.2 | |||
| 2aPC | −31.9 | −33.6 | −41.5 | −42.7 | |||
| 2bPC | −42.4 | −44.6 | −51.8 | −50.9 | |||
| 2cPC | −44.2 | −41.6 | −49.0 | −54.0 | |||
| 2dPC | −42.4 | −44.6 | −51.8 | −54.0 | |||
| 2P1 | −15.5 | −18.1 | −22.5 | −26.3 | −26.2 | −37.8 | |
| 2P2 | −31.3 | −34.0 | −32.0 | −42.1 | −44.0 | −48.7 | |
| iC3H7 | 3a/3bRC | −20.7 | −21.4 | −21.9 | −22.8 | ||
| 3c/3dRC | −15.5 | −16.3 | −16.9 | −17.9 | |||
| 3aTS | −11.6 | −12.4 | −16.9 | −17.5 | |||
| 3bTS | −9.1 | −11.0 | −14.1 | −16.3 | |||
| 3cTS | 0.3 | −1.0 | −4.3 | −5.4 | |||
| 3dTS | 31.5 | 28.8 | 21.4 | - | |||
| 3aPC | −31.8 | −33.8 | −41.8 | −44.4 | |||
| 3bPC | −43.9 | −46.9 | −54.0 | −57.9 | |||
| 3cPC | −44.4 | −47.3 | −48.8 | −52.8 | |||
| 3dPC | −44.4 | −47.3 | −54.0 | - | |||
| 3P1 | −13.9 | −16.5 | −22.1 | −24.8 | −28.4 | −37.7 | |
| 3P2 | −31.8 | −34.9 | −30.3 | −42.8 | −46.8 | −45.9 | |
| tC4H9 | 4a/4bRC | −23.1 | −23.3 | −24.4 | −25.6 | ||
| 4c/4dRC | −15.6 | −16.1 | −17.2 | −17.8 | |||
| 4aTS | −11.9 | −17.6 | −17.7 | −24.4 | |||
| 4bTS | −2.3 | - | −7.5 | - | |||
| 4cTS | 0.8 | −4.6 | −3.9 | −10.8 | |||
| 4dTS | 39.9 | - | 28.8 | - | |||
| 4aPC | −33.4 | −37.8 | −43.4 | −51.2 | |||
| 4bPC | −46.2 | - | −56.1 | - | |||
| 4cPC | −44.0 | −49.1 | −49.1 | −57.4 | |||
| 4dPC | −46.2 | - | −56.1 | - | |||
| 4P1 | −12.6 | −18.2 | −22.1 | −23.9 | −37.6 | −37.6 | |
| 4P2 | −31.9 | - | −29.5 | −43.3 | - | −37.8 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhen, W.; Zhao, S.; Fu, G.; Wang, H.; Sun, J.; Yang, L.; Zhang, J. Effects of Methyl Substitution and Leaving Group on E2/SN2 Competition for Reactions of F− with RY (R = CH3, C2H5, iC3H7, tC4H9; Y = Cl, I). Molecules 2023, 28, 6269. https://doi.org/10.3390/molecules28176269
Zhen W, Zhao S, Fu G, Wang H, Sun J, Yang L, Zhang J. Effects of Methyl Substitution and Leaving Group on E2/SN2 Competition for Reactions of F− with RY (R = CH3, C2H5, iC3H7, tC4H9; Y = Cl, I). Molecules. 2023; 28(17):6269. https://doi.org/10.3390/molecules28176269
Chicago/Turabian StyleZhen, Wenqing, Siwei Zhao, Gang Fu, Hongyi Wang, Jianmin Sun, Li Yang, and Jiaxu Zhang. 2023. "Effects of Methyl Substitution and Leaving Group on E2/SN2 Competition for Reactions of F− with RY (R = CH3, C2H5, iC3H7, tC4H9; Y = Cl, I)" Molecules 28, no. 17: 6269. https://doi.org/10.3390/molecules28176269