

Recent Progresses in the Catalytic Stereoselective Dearomatization of Pyridines

Abstract
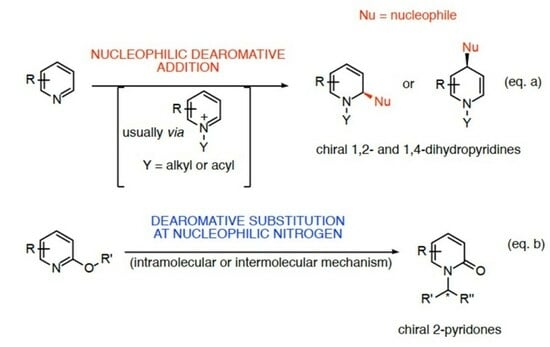
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Catalytic Stereoselective Dearomatization of Pyridine Scaffolds with Nucleophiles
2.1. Metal Catalysts
2.1.1. Grignard Reagents
2.1.2. Boronic Acids
2.1.3. Alkenes
2.1.4. Miscellaneous
2.2. Organocatalysis
3. Dearomative Substitution at Nucleophilic Pyridine Nitrogen
3.1. Intramolecular Dearomative Reactions of 2-Substituted Pyridines
3.2. Intermolecular Dearomative Reactions of (2-Hydroxy)Pyridines
3.3. Intermolecular Reactions of 4-Hydroxypyridines
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ding, Q.; Zhou, X.; Fan, R. Recent advances in dearomatization of heteroaromatic compounds. Org. Biomol. Chem. 2014, 12, 4807–4815. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; You, S.-L. Advances in Catalytic Asymmetric Dearomatization. ACS Cent. Sci. 2021, 7, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–11027. [Google Scholar] [CrossRef] [PubMed]
- Comins, D.L.; Higuchi, K.; Young, D.W. Dihydropyridine Preparation and Application in the Synthesis of Pyridine Derivatives. Adv. Heterocycl. Chem. 2013, 110, 175. [Google Scholar]
- Menichetti, A.; Berti, F.; Pineschi, M. Nitroso Diels-Alder cycloadducts derived from N-acyl-1,2-dihydropyridines as a new platform to molecular diversity. Molecules 2020, 25, 563. [Google Scholar] [CrossRef]
- Berti, F.; Menichetti, A.; Favero, L.; Marchetti, F.; Pineschi, M. Regio- and stereodivergent allylic reductions of bicyclic piperidine enecarbamate derivatives. J. Org. Chem. 2018, 83, 12221–12228. [Google Scholar] [CrossRef]
- Berti, F.; Menichetti, A.; Di Bussolo, V.; Favero, L.; Pineschi, M. Synthesis of bicyclic piperidinyl enamides and enecarbamates by hetero-Cope rearrangement of nitroso cycloadducts. Chem. Heterocycl. Compd. 2018, 54, 458–468. [Google Scholar] [CrossRef]
- Shang, Y.; Wu, C.; Gao, Q.; Liu, C.; Li, L.; Zhang, X.; Cheng, H.-G.; Liu, S.; Zhou, Q. Diversity-oriented functionalization of 2-pyridones and uracils. Nat. Commun. 2021, 12, 2988. [Google Scholar] [CrossRef]
- Wu, Y.-B.; Wu, Y.-Z.; Wu, J.; Jiang, H.; Chang, W.-W.; Ma, C.-Y. Copper-Catalyzed Regioselective Coupling of Tosylhydrazones and 2-Pyridones: A Strategy for the Production of N-alkylated Compounds. J. Org. Chem. 2021, 86, 6918–6926. [Google Scholar] [CrossRef]
- Bull, J.A.; Mosseau, J.J.; Pelletier, J.; Charette, A.B. Synthesis of pyridine and dihydropyridine derivatives by regio- and stereoselective addition to N-activated pyridines. Chem. Rev. 2012, 112, 2642–2713. [Google Scholar] [CrossRef]
- Bertuzzi, G.; Bernardi, L.; Fochi, M. Nucleophilic Dearomatization of Activated Pyridines. Catalysts 2018, 8, 632. [Google Scholar] [CrossRef]
- Wagener, T.; Lüchemeier, L.; Daniliuc, C.G.; Glorius, F. Interrupted Pyridine Hydrogenation: Asymmetric Synthesis of δ-Lactams. Angew. Chem. Int. Ed. 2021, 60, 6425–6429. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.; Barnard, J.H.; Gunasekar, R.; Pu, C.; Wu, X.; Zhang, S.; Ruan, J.; Xiao, J. Synthesis of chiral piperidines from pyridinium salts via rhodium-catalysed transfer hydrogenation. Nat. Catal. 2022, 5, 982–992. [Google Scholar] [CrossRef]
- Glorius, F. Asymmetric hydrogenation of aromatic compounds. Org. Biomol. Chem. 2005, 3, 4171–4175. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.G. Asymmetric Hydrogenation of Heteroaromatic Compounds. Acc. Chem. Res. 2007, 40, 1357–1366. [Google Scholar] [CrossRef]
- Yu, J.; Shi, F.; Gong, L.-Z. Brønsted-Acid-Catalyzed Asymmetric Multicomponent Reactions for the Facile Synthesis of Highly Enantioenriched Structurally Diverse Nitrogenous Heterocycles. Acc. Chem. Res. 2011, 44, 1156–1171. [Google Scholar] [CrossRef]
- Thu Pham, H.; Chataigner, I.; Renaud, J.L.; New approaches to nitrogen-containing heterocycles. Enantioselective organocatalyzed synthesis of dihydropyridines (DHP’s), quinolizidine derivatives and dihydropyrimidines (DHPM’s). Curr. Org. Chem. 2012, 16, 1754–1775. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, R. Recent Developments in Catalytic Asymmetric Inverse-Electron-Demand Diels–Alder Reaction. Chem. Rev. 2013, 113, 5515–5546. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.T.; Zhang, L.; You, S.L. Catalytic asymmetric dearomatization (CADA) reactions of phenol and aniline derivatives. Chem. Soc. Rev. 2016, 45, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, E.; Suzuki, M.; Yabu, K.; Albert, M.; Kanai, M.; Shibasaki, M. New Entries in Lewis Acid–Lewis Base Bifunctional Asymmetric Catalyst: Catalytic Enantioselective Reissert Reaction of Pyridine Derivatives. J. Am. Chem. Soc. 2004, 126, 11808–11809. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Yu, S.; Ding, Z.; Ma, D. Enantioselective Addition of Activated Terminal Alkynes to 1-Acylpyridinium Salts Catalyzed by Cu–Bis(oxazoline) Complexes. J. Am. Chem. Soc. 2007, 129, 9300–9301. [Google Scholar] [CrossRef]
- Black, D.A.; Beveridge, R.E.; Arndtsen, B.A. Copper-Catalyzed Coupling of Pyridines and Quinolines with Alkynes: A One-Step, Asymmetric Route to Functionalized Heterocycles. J. Org. Chem. 2008, 73, 1906–1910. [Google Scholar] [CrossRef]
- Christian, N.; Aly, S.; Belyk, K. Rhodium-Catalyzed Enantioselective Addition of Boronic Acids to N-Benzylnicotinate Salts. J. Am. Chem. Soc. 2011, 133, 2878–2880. [Google Scholar] [PubMed]
- Lutz, J.P.; Chau, S.T.; Doyle, A.G. Nickel-catalyzed enantioselective arylation of pyridine. Chem. Sci. 2016, 7, 4105–4109. [Google Scholar] [CrossRef]
- García Mancheño, O.; Asmus, S.; Zurro, M.; Fischer, T. Highly Enantioselective Nucleophilic Dearomatization of Pyridines by Anion-Binding Catalysis. Angew. Chem. Int. Ed. 2015, 54, 8823–8827. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, G.; Sinisi, A.; Caruana, L.; Mazzanti, A.; Fochi, M.; Bernardi, L. Catalytic Enantioselective Addition of Indoles to Activated N-Benzylpyridinium Salts: Nucleophilic Dearomatization of Pyridines with Unusual C-4 Regioselectivity. ACS Catal. 2016, 6, 6473–6477. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Rovis, T. Enantioselective N-heterocyclic carbene-catalyzed nucleophilic dearomatization of alkyl pyridiniums. Chem. Sci. 2017, 8, 6566–6569. [Google Scholar] [CrossRef] [PubMed]
- Di Carmine, G.; Ragno, D.; Bortolini, O.; Giovannini, P.P.; Mazzanti, A.; Massi, A.; Fogagnolo, M. Enantioselective Dearomatization of Alkylpyridiniums by N-Heterocyclic Carbene-Catalyzed Nucleophilic Acylation. J. Org. Chem. 2018, 83, 2050–2057. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, G.; Sinisi, A.; Pecorari, D.; Caruana, L.; Mazzanti, A.; Bernardi, L.; Fochi, M. Nucleophilic Dearomatization of Pyridines under Enamine Catalysis: Regio-, Diastereo-, and Enantioselective Addition of Aldehydes to Activated N-Alkylpyridinium Salts. Org. Lett. 2017, 19, 834–837. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Z.; Liu, Z.; Huang, M.; Hu, J.; Yu, P. Strategic C-C Bond-Forming Dearomatization of Pyridines and Quinolines. Org. Lett. 2019, 21, 4459–4463. [Google Scholar] [CrossRef]
- Guo, Y.; Reis, M.C.; Kootstra, J.; Harutyunyan, S.R. Enantioselective Catalytic Dearomative Addition of Grignard Reagents to 4-Methoxypyridinium Ions. ACS Catal. 2021, 11, 8476–8483. [Google Scholar] [CrossRef] [PubMed]
- Somprasong, S.; Reis, M.C.; Harutyunyan, S.R. Catalytic Access to Chiral δ-Lactams via Nucleophilic Dearomatization of Pyridine Derivatives. Angew. Chem. Int. Ed. 2023, 62, e202217328. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, E.I.; Hergenrother, P.J. Runaway ROS as a Selective Anticancer Strategy. ChemMedChem 2011, 6, 1957–1959. [Google Scholar] [CrossRef] [PubMed]
- Bezerra, D.P.; Pessoa, C.; Odorico de Moraes, M.; Saker-Neto, N.; Silveira, E.R.; Costa-Lotufo, L.V. Overview of the therapeutic potential of piplartine (piperlongumine). Eur. J. Pharm. Sci. 2013, 48, 453–463. [Google Scholar] [CrossRef]
- Peng, S.; Zhang, B.; Meng, X.; Yao, J.; Fang, J. Synthesis of Piperlongumine Analogues and Discovery of Nuclear Factor Erythroid 2-Related Factor 2 (Nrf2) Activators as Potential Neuroprotective Agents. J. Med. Chem. 2015, 58, 5242–5255. [Google Scholar] [CrossRef]
- Wilde, J.H.; Dickie, D.A.; Harman, W.D. A Highly Divergent Synthesis of 3-Aminotetrahydropyridines. J. Org. Chem. 2020, 85, 8245–8252. [Google Scholar] [CrossRef]
- Wilson, K.B.; Smith, J.A.; Nedzbala, H.S.; Pert, E.K.; Dakermanji, S.J.; Dickie, D.A.; Harman, W.D. Highly Functionalized Cyclohexenes Derived from Benzene: Sequential Tandem Addition Reactions Promoted by Tungsten. J. Org. Chem. 2019, 84, 6094–6116. [Google Scholar] [CrossRef]
- Lankenau, A.W.; Iovan, D.A.; Pienkos, J.A.; Salomon, R.J.; Wang, S.; Harrison, D.P.; Myers, W.H.; Harman, W.D. Enantioenrichment of a Tungsten Dearomatization Agent Utilizing Chiral Acids. J. Am. Chem. Soc. 2015, 137, 3649–3655. [Google Scholar] [CrossRef]
- Graham, P.M.; Delafuente, D.A.; Liu, W.; Myers, W.H.; Sabat, M.; Harman, W.D. Facile Diels-Alder Reactions with Pyridines Promoted by Tungsten. J. Am. Chem. Soc. 2005, 127, 10568–10572. [Google Scholar] [CrossRef]
- Grigolo, T.A.; Subhit, A.R.; Smith, J.M. Regioselective Asymmetric Alkynylation of N-Alkyl Pyridiniums. Org. Lett. 2021, 23, 6703–6708. [Google Scholar] [CrossRef]
- Hashimoto, S.; Yamada, S.; Koga, K. Asymmetric Syntheses Using Tert-Leucine. 1. An Asymmetric Synthesis of Beta-Substituted Aldehydes via 1,4-Addition of Grignard Reagents to Chiral Alpha,Beta-Unsaturated Aldimines. J. Am. Chem. Soc. 1976, 98, 7450–7452. [Google Scholar] [CrossRef] [PubMed]
- Grigolo, T.A.; Smith, J.M. Regiodivergent Asymmetric Pyridinium Additions: Mechanistic Insight and Synthetic Applications. Chem. Eur. J. 2022, 28, e202202813. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.J.; Spurlin, S.P.; Gorden, J.D.; Karimov, R.R. Enantioselective Synthesis of Dihydropyridines Containing Quaternary Stereocenters Through Dearomatization of Pyridinium Salts. ACS Catal. 2020, 10, 51–55. [Google Scholar] [CrossRef]
- Gribble, M.W., Jr.; Guo, S.; Buchwald, S.L. Asymmetric Cu-Catalyzed 1,4-Dearomatization of Pyridines and Pyridazines without Preactivation of the Heterocycle or Nucleophile. J. Am. Chem. Soc. 2018, 140, 5057–5060. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Sheong, F.K.; Lin, Z. DFT Studies on Copper-Catalyzed Dearomatization of Pyridine. ACS Catal. 2020, 10, 9585–9593. [Google Scholar] [CrossRef]
- Gribble, M.W., Jr.; Liu, R.Y.; Buchwald, S.L. Evidence for Simultaneous Dearomatization of Two Aromatic Rings under Mild Conditions in Cu(I)-Catalyzed Direct Asymmetric Dearomatization of Pyridine. J. Am. Chem. Soc. 2020, 142, 11252–11269. [Google Scholar] [CrossRef]
- Mishra, S.; Karabiyikoglu, S.; Fletcher, S.P. Catalytic Enantioselective Synthesis of 3-Piperidines from Arylboronic Acids and Pyridine. J. Am. Chem. Soc. 2023, 145, 14221–14226. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.-J.; Xiao, B.-X.; Ouyang, Q.; Liang, H.-P.; Du, W.; Chen, Y.-C. Asymmetric Dearomative Formal [4 + 2] Cycloadditions of N,4-Dialkylpyridinium Salts and Enones to Construct Azaspiro [5.5]undecane Frameworks. Org. Lett. 2018, 20, 8000–8003. [Google Scholar] [CrossRef]
- Song, X.; Yan, R.-J.; Du, W.; Chen, Y.-C. Asymmetric Dearomative Cascade Multiple Functionalizations of Activated N-Alkylpyridinium and N-Alkylquinolinium Salts. Org. Lett. 2020, 22, 7617–7621. [Google Scholar] [CrossRef]
- McLaughlin, C.; Bitai, J.; Barber, L.J.; Slawin, A.M.Z.; Smith, A.D. Catalytic enantioselective synthesis of 1,4-dihydropyridines via the addition of C(1)-ammonium enolates to pyridinium salts. Chem. Sci. 2021, 12, 12001–12011. [Google Scholar] [CrossRef]
- Pan, G.; He, C.; Chen, M.; Xiong, Q.; Cao, W.; Feng, X. Synthesis of Dihydroisoquinoline and Dihydropyridine Derivatives via Asymmetric Dearomative Three-Component Reaction. CCS Chem. 2022, 4, 2000–2008. [Google Scholar] [CrossRef]
- Kerkovius, J.K.; Stegner, A.; Turlik, A.; Lam, P.H.; Houk, K.N.; Reisman, S.E. A Pyridine Dearomatization Approach to the Matrine-Type Lupin Alkaloids. J. Am. Chem. Soc. 2022, 144, 15938–15943. [Google Scholar] [CrossRef] [PubMed]
- Harawa, V.; Thorpe, T.W.; Marshall, J.R.; Sangster, J.J.; Gilio, A.K.; Pirvu, L.; Heath, R.S.; Angelastro, A.; Finnigan, J.D.; Charnock, S.J.; et al. Synthesis of stereoenriched piperidines via chemo-enzymatic dearomatization of activated pyridines. J. Am. Chem. Soc. 2022, 144, 21088–21095. [Google Scholar] [CrossRef]
- Pfefferkorn, J.A.; Lou, J.; Minich, M.L.; Filipski, K.J.; He, M.; Zhou, R.; Ahmed, S.; Benbow, J.; Perez, A.-G.; Tu, M.; et al. Pyridones as glucokinase activators: Identification of a unique metabolic liability of the 4-sulfonyl-2-pyridone heterocycle. Bioorg. Med. Chem. Lett. 2009, 19, 3247–3252. [Google Scholar] [CrossRef] [PubMed]
- Straub, C.S.; Padwa, A. Synthesis of the Angiotensin Converting Enzyme Inhibitor(−)-A58365A via an Isomunchnone Cycloaddition Reaction. Org. Lett. 1999, 1, 83–85. [Google Scholar] [CrossRef]
- Clive, D.L.J.; Coltart, D.M.; Zhou, Y. Synthesis of the Angiotensin-Converting Enzyme Inhibitors (−)-A58365A and (−)-A58365B from a Common Intermediate. J. Org. Chem. 1999, 64, 1447–1454. [Google Scholar] [CrossRef]
- Moeller, K.D.; Wong, P.L. Anodic Amide Oxidation: A Novel Synthesis of the Angiotensin Converting Enzyme Inhibitor A58365A. Bioorg. Med. Chem. Lett. 1992, 2, 739–742. [Google Scholar] [CrossRef]
- Fang, F.G.; Danishefsky, S.J. Total synthesis of the angiotensin-converting enzyme inhibitor A58365A: On the use of pyroglutamate as a chiral educt. Tetrahedron Lett. 1989, 30, 3621–3624. [Google Scholar] [CrossRef]
- Rodrigues, A.; Lee, E.E.; Batey, R.A. Enantioselective Palladium(II)-Catalyzed Formal Rearrangement of 2-Allyloxypyridines and Related Heterocycles. Org. Lett. 2010, 12, 260–263. [Google Scholar] [CrossRef]
- Yeung, C.S.; Hsieh, T.H.H.; Dong, V.M. Ru-catalyzed activation of sp3 C-O bonds: O- to N-alkyl migratory rearrangements in pyridines and related heterocycles. Chem. Sci. 2011, 2, 544–551. [Google Scholar] [CrossRef]
- Pan, S.; Ryu, N.; Shibata, T. Ir(I)-Catalyzed Synthesis of N-Substituted Pyridones from 2-Alkoxypyridines via C–O Bond Cleavage. Org. Lett. 2013, 15, 1902–1905. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.-J.; Brown, A.P.N.; Cordier, C.J. Enantioselective propargylic [1,3]-rearrangements: Copper-catalyzed O- to N migrations towards C-N bond formation. Chem. Sci. 2017, 8, 4299–4305. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.P.; Wu, Q.-F.; You, S.-L. Direct Asymmetric Dearomatization of Pyridines and Pyrazines by Iridium-Catalyzed Allylic Amination reactions. Angew. Chem. Int. Ed. 2014, 53, 6986–6989. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.-P.; Wu, Q.-F.; Shao, W.; You, S.-L. Iridium-Catalyzed Intramolecular Asymmetric Allylic Dearomatization Reaction of Pyridines, Pyrazines, Quinolines, and Isoquinolines. J. Am. Chem. Soc. 2015, 137, 15899–15906. [Google Scholar] [CrossRef]
- Min, X.-L.; Zhang, X.-L.; Yi, W.; He, Y. Brønsted acid-enhanced copper-catalyzed atroposelective cycloisomerization to axially chiral arylquinolizones via dearomatization of pyridine. Nat. Commun. 2022, 13, 373. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, T.P.; Dutta, I.; Huang, K.-W. Aromaticity in catalysis: Metal ligand cooperation via ligand dearomatization and rearomatization. Chem. Commun. 2021, 57, 3070–3082. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Kähny, M.; Breit, B. Rhodium-Catalyzed Chemo-, Regio-, and Enantioselective Addition of 2-Pyridones to Terminal Allenes. Angew. Chem. Int. Ed. 2014, 53, 13780–13784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, Z.-P.; Huang, L.; You, S.-L. Highly Regio- and Enantioselective Synthesis of N-substituted 2-Pyridones: Iridium-Catalyzed Intermolecular Asymmetric Allylic Amination. Angew. Chem. Int. Ed. 2015, 54, 1873–1876. [Google Scholar] [CrossRef]
- Sun, C.; Qi, X.; Min, X.-L.; Bai, X.-L.; Liu, P.; He, Y. Asymmetric allylic substitution-isomerization to axially chiral enamides via hydrogen-bonding assisted central-to-axial chirality transfer. Chem. Sci. 2020, 11, 10119–10126. [Google Scholar] [CrossRef]
- Khan, S.; Shah, B.H.; Kan, I.; Li, M.; Zhang, Y.J. Pd-catalyzed regio- and enantioselective allylic substitution with 2-pyridones. Chem. Commun. 2019, 55, 13168–13171. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Y.; Khan, S.; Zhang, Z.; Khan, A. Selective approach to N-substituted tertiary 2-pyridones. New J. Chem. 2022, 46, 11138–11142. [Google Scholar] [CrossRef]
- Yao, K.; Yuan, Q.; Qu, X.; Liu, Y.; Liu, D.; Zhang, W. Pd-catalyzed asymmetric allylic substitution cascade using α-(pyridine-1-yl)-acetamides formed in situ as nucleophiles. Chem. Sci. 2019, 10, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chen, P.; Liu, P.; Tang, S.; Zhang, X.; Sun, J. Access to N-Substituted 2-Pyridones by Catalytic Intermolecular Dearomatization and 1,4-Acyl Transfer. Angew. Chem. Int. Ed. 2019, 58, 1980–1984. [Google Scholar] [CrossRef]
- Wang, K.; Liu, Z.; Xu, G.; Shao, Y.; Tang, S.; Chen, P.; Zhang, X.; Sun, J. Chemo- and Enantioselective Insertion of Furyl Carbene into the N-H bond of 2-Pyridones. Angew. Chem. Int. Ed. 2021, 60, 16942–16946. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-C.; Jhong, Y.; Lin, H.-J.; Swain, S.P.; Tsai, H.-H.G.; Hou, D.-R. Organocatalyzed enantioselective Michael addition of 2-hydroxypyridines and α,β-unsaturated 1,4-dicarbonyl compounds. Adv. Synth. Catal. 2019, 361, 4966–4982. [Google Scholar] [CrossRef]
- Shao, W.; Wang, Y.; Yang, Z.-P.; Zhang, X.; You, S.-L. Efficient Synthesis of N-Alkylated 4-Pyridones by Copper-Catalyzed Intermolecular Asymmetric Propargylic Amination. Chem. Asian. J. 2018, 13, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.P.; Li, C.; Breit, B. Transition-Metal-Catalyzed Regiodivergent and Stereoselective Access to Branched and Linear Allylated 4-Pyridones. Chem. Eur. J. 2017, 23, 6531–6534. [Google Scholar] [CrossRef]
- Tu, H.-F.; Nie, Y.-H.; Zheng, C.; You, S.-L. Iridium-Catalyzed Intermolecular Asymmetric Allylic Amination with Pyridones. Adv. Synth. Catal. 2022, 364, 3432–3437. [Google Scholar] [CrossRef]


































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comparini, L.M.; Pineschi, M. Recent Progresses in the Catalytic Stereoselective Dearomatization of Pyridines. Molecules 2023, 28, 6186. https://doi.org/10.3390/molecules28176186
Comparini LM, Pineschi M. Recent Progresses in the Catalytic Stereoselective Dearomatization of Pyridines. Molecules. 2023; 28(17):6186. https://doi.org/10.3390/molecules28176186
Chicago/Turabian StyleComparini, Lucrezia Margherita, and Mauro Pineschi. 2023. "Recent Progresses in the Catalytic Stereoselective Dearomatization of Pyridines" Molecules 28, no. 17: 6186. https://doi.org/10.3390/molecules28176186