
Isolation and Identification of Phytocompounds from Maytenus dhofarensis and Their Biological Potentials
 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
3. Experimental Section
3.1. General Experimental Procedures
Solvents and Reagents
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Computational Studies of Compounds 1 and 3
3.5. Biological Assays
3.5.1. Homogenous Time-Resolved Fluorescence (HTRF) Kinase Assay
3.5.2. Antioxidant Assay Activity Using 2,2′-Diphenyl-1-picrylhydrazyl (DPPH) Radical-Scavenging Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Simmons, M.P.; Savolainen, V.; Clevinger, C.C.; Archer, R.H.; Davis, J.I. Phylogeny of the Celastraceae inferred from 26S nuclear ribosomal DNA, phytochrome B, rbcL, atpB, and morphology. Mol. Phylogenetics Evol. 2001, 19, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, J.G.; Delle Monache, G.; Delle Monache, F.; Marini-Bettolo, G.B. Chuchuhuasha—A drug used in folk medicine in the Amazonian and Andean areas. A chemical study of Maytenus laevis. J. Ethnopharmacol. 1982, 5, 73–77. [Google Scholar] [CrossRef]
- Veloso, C.; Oliveira, M.; Rodrigues, V.; Oliveira, C.; Duarte, L.; Teixeira, M.; Ferreira, A.; Perez, A. Evaluation of the effects of extracts of Maytenus imbricata (Celastraceae) on the treatment of inflammatory and metabolic dysfunction induced by high-refined carbohydrate diet. Inflammopharmacology 2019, 27, 539–548. [Google Scholar] [PubMed]
- Malaník, M.; Treml, J.; Rjašková, V.; Tížková, K.; Kaucká, P.; Kokoška, L.; Kubatka, P.; Šmejkal, K. Maytenus macrocarpa (Ruiz & Pav.) Briq.: Phytochemistry and Pharmacological Activity. Molecules 2019, 24, 2288. [Google Scholar] [PubMed] [Green Version]
- de Figueiredo, P.T.; Silva, E.W.; Cordeiro, L.V.; Barros, R.P.; Lima, E.; Scotti, M.T.; da Silva, M.S.; Tavares, J.F.; Costa, V.C.d.O. Lupanes and friedelanes, the first chemical constituents of the aerial parts of Maytenus erythroxylon Reissek. Phytochem. Lett. 2021, 45, 19–24. [Google Scholar] [CrossRef]
- Taddeo, V.A.; Castillo, U.G.; Martínez, M.L.; Menjivar, J.; Jiménez, I.A.; Núñez, M.J.; Bazzocchi, I.L. Development and validation of an HPLC-PDA method for biologically active quinonemethide triterpenoids isolated from Maytenus chiapensis. Medicines 2019, 6, 36. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Ji, M.-Y.; Qiu, B.; Li, Q.-Y.; Zhang, K.-Y.; Liu, J.-C.; Dang, L.-S.; Li, M.-H. Phytochemicals and biological activities of species from the genus Maytenus. Med. Chem. Res. 2020, 29, 575–606. [Google Scholar] [CrossRef]
- De Sousa, G.F.; de Aguilar, M.G.; Takahashi, J.A.; Alves, T.M.; Kohlhoff, M.; Vieira Filho, S.A.; Silva, G.D.; Duarte, L.P. Flavonol triglycosides of leaves from Maytenus robusta with acetylcholinesterase inhibition. Phytochem. Lett. 2017, 19, 34–38. [Google Scholar]
- Pino, L.L.; García, T.H.; Delgado-Roche, L.; Rodeiro, I.; Hernández, I.; Vilegas, W.; Spengler, I. Polyphenolic profile by FIA/ESI/IT/MSn and antioxidant capacity of the ethanolic extract from the barks of Maytenus cajalbanica (Borhidi & O. Muñiz) Borhidi & O. Muñiz. Nat. Prod. Res. 2020, 34, 1481–1485. [Google Scholar]
- Olivaro, C.; Escobal, M.; de Souza, G.; Mederos, A. Chemical characterisation and in vitr o anthelmintic activity of phenolic-rich extracts from the leaves and branches of Maytenus ilicifolia, a native plant from South America. Nat. Prod. Res. 2022, 36, 3168–3172. [Google Scholar] [CrossRef]
- Vazdekis, N.E.; Chavez, H.; Estevez-Braun, A.; Ravelo, A.G. Triterpenoids and a lignan from the aerial parts of Maytenus apurimacensis. J. Nat. Prod. 2009, 72, 1045–1048. [Google Scholar] [CrossRef]
- Okoye, F.B.C.; Agbo, M.O.; Nworu, C.S.; Nwodo, N.J.; Esimone, C.O.; Osadebe, P.O.; Proksch, P. New neolignan glycoside and an unusual benzoyl malic acid derivative from Maytenus senegalensis leaves. Nat. Prod. Res. 2015, 29, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Perestelo, N.R.; Jiménez, I.A.; Tokuda, H.; Vázquez, J.T.; Ichiishi, E.; Bazzocchi, I.L. Absolute configuration of dihydro-β-agarofuran sesquiterpenes from Maytenus jelskii and their potential antitumor-promoting effects. J. Nat. Prod. 2016, 79, 2324–2331. [Google Scholar] [CrossRef] [PubMed]
- Alarcón-Enos, J.; Muñoz-Núñez, E.; Gutiérrez, M.; Quiroz-Carreño, S.; Pastene-Navarrete, E.; Céspedes Acuña, C. Dyhidro-β-agarofurans natural and synthetic as acetylcholinesterase and COX inhibitors: Interaction with the peripheral anionic site (AChE-PAS), and anti-inflammatory potentials. J. Enzyme Inhib. Med. Chem. 2022, 37, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Núñez, M.J.; Guadaño, A.; Jiménez, I.A.; Ravelo, A.G.; González-Coloma, A.; Bazzocchi, I.L. Insecticidal Sesquiterpene Pyridine Alkaloids from Maytenus c hiapensis. J. Nat. Prod. 2004, 67, 14–18. [Google Scholar] [CrossRef]
- Santos, V.A.F.F.M.; Regasini, L.O.; Nogueira, C.u.R.; Passerini, G.D.; Martinez, I.; Bolzani, V.S.; Graminha, M.r.A.; Cicarelli, R.M.; Furlan, M. Antiprotozoal sesquiterpene pyridine alkaloids from Maytenus ilicifolia. J. Nat. Prod. 2012, 75, 991–995. [Google Scholar] [CrossRef]
- Din, A.U.; Siddiqui, B.S. Royleanine A, an Antitumor Dihydro-β-agarofuran Sesquiterpene Pyridine Alkaloid from Maytenus royleanus. J. Braz. Chem. Soc. 2022, 33, 1386–1391. [Google Scholar]
- Wang, C.; Li, C.-J.; Yang, J.-Z.; Ma, J.; Chen, X.-G.; Hou, Q.; Zhang, D.-M. Anti-inflammatory sesquiterpene derivatives from the leaves of Tripterygium wilfordii. J. Nat. Prod. 2013, 76, 85–90. [Google Scholar] [CrossRef]
- González, A.G.; Jiménez, I.A.; Ravelo, A.G.; Sazatornil, J.G.; Bazzocchi, I.L. New sesquiterpenes with antifeedant activity from Maytenus canariensis (Celastraceae). Tetrahedron 1993, 49, 697–702. [Google Scholar] [CrossRef]
- Wibowo, M.; Wang, Q.; Holst, J.; White, J.M.; Hofmann, A.; Davis, R.A. Dihydro-β-agarofurans from the roots of the Australian endemic rainforest tree Maytenus bilocularis act as leucine transport inhibitors. Phytochem 2018, 148, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Touré, S.; Nirma, C.; Falkowski, M.; Dusfour, I.; Boulogne, I.; Jahn-Oyac, A.; Coke, M.; Azam, D.; Girod, R.; Moriou, C. Aedes aegypti larvicidal sesquiterpene alkaloids from Maytenus oblongata. J. Nat. Prod. 2017, 80, 384–390. [Google Scholar] [PubMed]
- Zhu, Y.D.; Miao, Z.H.; Ding, J.; Zhao, W.M. Cytotoxic Dihydroagarofuranoid Sesquiterpenes from the Seeds of Celastrus orbiculatus. J. Nat. Prod. 2008, 71, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.J.; Wang, M.G.; Zhu, J.B.; Zhou, W.M.; Hu, Z.N.; Ji, Z.Q. Five New Insecticidal Sesquiterpenoids from Celastrus angulatus. J. Nat. Prod. 2001, 64, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Bazzocchi, I.L.; Nunez, M.J.; Pardo, L.; Castanys, S.; Campillo, M.; Jimenez, I.A. Biological Evaluation, Structure–Activity Relationships, and Three-Dimensional Quantitative Structure–Activity Relationship Studies of Dihydro-β-agarofuran Sesquiterpenes as Modulators of P-Glycoprotein-Dependent Multidrug Resistance. J. Med. Chem. 2007, 50, 4808–4817. [Google Scholar]
- Callies, O.; Sanchez-Canete, M.P.; Gamarro, F.; Jimenez, I.A.; Castanys, S.; Bazzocchi, I.L. Restoration of Chemosensitivity in P-Glycoprotein-Dependent Multidrug-Resistant Cells by Dihydro-β-agarofuran Sesquiterpenes from Celastrus vulcanicola. J. Nat. Prod. 2015, 78, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Callies, O.; Sanchez-Canete, M.P.; Gamarro, F.; Jimenez, I.A.; Castanys, S.; Bazzocchi, I.L. Optimization by Molecular Fine Tuning of Dihydro-β-agarofuran Sesquiterpenoids as Reversers of P-Glycoprotein-Mediated Multidrug Resistance. J. Med. Chem. 2016, 59, 1880–1890. [Google Scholar]
- Chen, J.J.; Chou, T.H.; Peng, C.F.; Chen, I.S.; Yang, S.Z. Antitubercular Dihydroagarofuranoid Sesquiterpenes from the Roots of Microtropis fokienensis. J. Nat. Prod. 2007, 70, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wang, W.; Gong, Q.; Zhang, H.; Zhao, W.M. Neuroprotective Dihydro-β-agarofuran-Type Sesquiterpenes from the Seeds of Euonymus maackii. J. Nat. Prod. 2019, 82, 3096–3103. [Google Scholar] [CrossRef]
- Luo, Y.; Pu, X.; Luo, G.; Zhou, M.; Ye, Q.; Liu, Y.; Gu, J.; Qi, H.; Li, G.; Zhang, G. Nitrogen-Containing Dihydro-β-agarofuran Derivatives from Tripterygium wilfordii. J. Nat. Prod. 2014, 77, 1650–1657. [Google Scholar]
- Gutierrez-Nicolas, F.; Oberti, J.C.; Ravelo, A.G.; EstevezBraun, A. β-Agarofurans and Sesquiterpene Pyridine Alkaloids from Maytenus spinosa. J. Nat. Prod. 2014, 77, 1853–1863. [Google Scholar]
- Corsino, J.; da Silva Bolzani, V.; Pereira, A.M.S.; França, S.C.; Furlan, M. Bioactive sesquiterpene pyridine alkaloids from Maytenus aquifolium. Phytochemistry 1998, 48, 137–140. [Google Scholar]
- Malebo, H.M.; Wiketye, V.; Katani, S.J.; Kitufe, N.A.; Nyigo, V.A.; Imeda, C.P.; Ogondiek, J.W.; Sunguruma, R.; Mhame, P.P.; Massaga, J.J. In vivo antiplasmodial and toxicological effect of Maytenus senegalensis traditionally used in the treatment of malaria in Tanzania. Malar. J. 2015, 14, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, G.; Serrano, R.; Silva, O. Maytenus heterophylla and Maytenus senegalensis, two traditional herbal medicines. J. Nat. Sci. Biol. Med. 2011, 2, 59. [Google Scholar]
- Abreu-Naranjo, R.; Arteaga-Crespo, Y.; Bravo-Sanchez, L.R.; Pérez-Quintana, M.L.; García-Quintana, Y. Response surface methodology for optimisation of total polyphenol content and antioxidant activity of extracts from Maytenus macrocarpa bark by means of ultrasound-assisted extraction. Wood Sci. Technol. 2018, 52, 1359–1376. [Google Scholar] [CrossRef]
- Niero, R.; Faloni de Andrade, S.; Cechinel Filho, V. A review of the ethnopharmacology, phytochemistry and pharmacology of plants of the Maytenus genus. Curr. Pharm. Des. 2011, 17, 1851–1871. [Google Scholar] [CrossRef] [PubMed]
- Veloso, C.C.; Soares, G.L.; Perez, A.C.; Rodrigues, V.G.; Silva, F.C. Pharmacological potential of Maytenus species and isolated constituents, especially tingenone, for treatment of painful inflammatory diseases. Rev. Bras. Farmacogn. 2017, 27, 533–540. [Google Scholar] [CrossRef]
- Moo-Puc, J.A.; Martín-Quintal, Z.; Mirón-López, G.; Moo-Puc, R.E.; Quijano, L.; Mena-Rejón, G.J. Isolation and antitrichomonal activity of the chemical constituents of the leaves of Maytenus phyllanthoides Benth. (Celastraceae). Quim. Nova 2014, 37, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Anthony, G.; Miller, M.M. Plants of Dhofar the Southern Region of Oman Traditional, Economic and Medicinal Uses; The Office of the Adviser for Conservation of the Environment, Diwan of Royal Court: Muscat, Oman, 1988; Volume 1. [Google Scholar]
- Grimblat, N.; Zanardi, M.M.; Sarotti, A.M. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision E.01.; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Degorce, F.; Card, A.; Soh, S.; Trinquet, E.; Knapik, G.P.; Xie, B. HTRF: A technology tailored for drug discovery–a review of theoretical aspects and recent applications. Curr. Chem. Genom. 2009, 3, 22. [Google Scholar] [CrossRef]
- Koul, H.K.; Pal, M.; Koul, S. Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 2013, 4, 342–359. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Fornace, A.J., Jr. p38 MAP kinase’s emerging role as a tumor suppressor. Adv. Cancer Res. 2004, 92, 95–118. [Google Scholar] [PubMed]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry; Oxford University Press Inc.: New York, NY, USA, 2012; p. 796. [Google Scholar]
- Braca, A.; De Tommasi, N.; Di Bari, L.; Pizza, C.; Politi, M.; Morelli, I. Antioxidant principles from bauhinia t arapotensis. J. Nat. Prod. 2001, 64, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Ferrigni, N.; McLaughlin, J.; Powell, R.; Smith, C., Jr. Use of potato disc and brine shrimp bioassays to detect activity and isolate piceatannol as the antileukemic principle from the seeds of Euphorbia lagascae. J. Nat. Prod. 1984, 47, 347–352. [Google Scholar] [CrossRef] [PubMed]
- MarvinView (Version 17.2.6.0) Developed by ChemAxon. 2017. Available online: http://www.chemaxon.com (accessed on 10 June 2023).
- Yuan-Yuan, H.; Lu, C.; Guo-Xu, M.; Xu-Dong, X.; Xue-Gong, J.; Fu-Sheng, D.; Xue-Jian, L.; Jing-Quan, Y. A Review on Phytochemicals of the Genus Maytenus and Their Bioactive Studies. Molecules 2021, 26, 4563. [Google Scholar]



{kind=link}
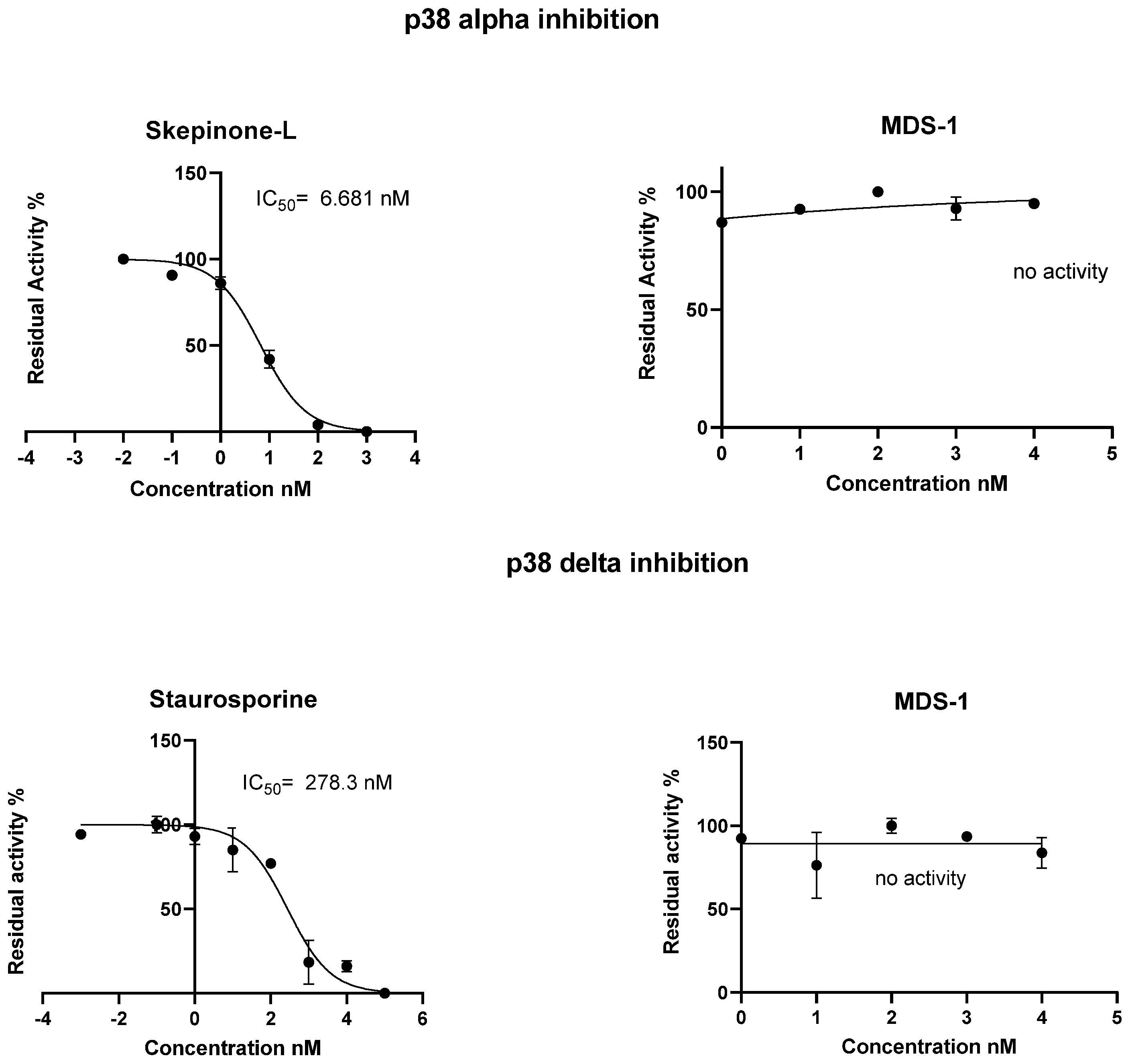{kind=link}
| Position | δC (ppm) | δH (ppm) (J in Hz) | HMBC (1H → 13C) |
|---|---|---|---|
| 1 | 70.6 | 5.44, d (3.2) | 2, 9′, 10, 15 |
| 2 | 68.3 | 5.53, m | - |
| 3 | 42.3 | Ha, 2.02, dd (3.2, 15.1) Hb, 2.18, dd (5.3, 16.0) | - |
| 4 | 69.9 | - | - |
| 5 | 91.1 | - | - |
| 6 | 78.7 | 6.22, s | 5, 7, 8, 10, 11, 16 |
| 7 | 49.2 | 2.35, m | - |
| 8 | 34.6 | Ha, 2.19, m Hb, 2.60, ddd (3.8, 11.1, 15.9) | - |
| 9 | 68.2 | 5.28, d (7.2) | 5, 7, 8, 10, 15 |
| 10 | 54.9 | - | - |
| 11 | 84.7 | - | - |
| 12 | 29.5 | 1.57, s | 7, 11, 13 |
| 13 | 25.7 | 1.52, s | 7, 11 |
| 14 | 24.9 | 1.49, s | 3, 5 |
| 15 | 65.5 | Ha, 4.92, d (12.8) Hb, 4.39, d (13.6) | 5, 9 5, 10 |
| 2` | 166.1 | - | - |
| 3` | 127.5 | - | - |
| 4` | 133.4 | 7.58, dd (7.4, 2.5) | 1′ |
| 5` | 128.7 | 7.48, dd (8.1, 7.6) | 3′ |
| 6` | 130.2 | 8.18, dd (8.3, 1.2) | 2′, 4′, 6′ |
| 7` | 140.2 | 6.93, dq (7.0, 1.3) | 2′, 10′, 11′ |
| 8` | 129.8 | - | - |
| 9` | 170.7 | - | - |
| 10` | 11.9 | 1.81, s | - |
| 11` | 14.7 | 1.85, s | 9` |
| OAc-6 | CH3: 20.6 | 1.78, s | - |
| C=O:169.5 | - | ||
| OAc-9 | CH3: 21.4 | 2.28, s | - |
| C=O:166.5 | - | - | |
| OAc-15 | CH3: 21.2 | 2.08, s | - |
| C=O:169.8 | - | - | |
| OH | - | 3.10 | 5, 7, 14 |
| Position | δC (ppm) | δH (ppm) (J in Hz) | HMBC (1H → 13C) |
|---|---|---|---|
| 1a | 38.5, CH2 | 0.98 a | |
| 1b | 1.61 a | ||
| 2 | 23.9, CH2 | 1.62 a, m | |
| 3 | 81.6, CH | 4.53, dd (14.0, 7.0) | 2, 4, 23, 24 |
| 4 | 37.9, C | ||
| 5 | 55.6, CH | 0.85 a | |
| 6 | 21.1, CH2 | 1.36 a | |
| 7 | 41.8, CH2 | 2.41, 2.39, dd (14.0, 7.0) | |
| 8 | 43.1, C | ||
| 9 | 50.5, CH | 1.29 a, m | |
| 10 | 37.2, C | ||
| 11 | 25.6, CH2 | 1.32 | |
| 12 | 27.6, CH2 | 0.88, t (7) | |
| 13 | 38.2, CH | 1.61 a, m | |
| 14 | 40.9, C | ||
| 15 | 34.3, CH2 | 1.35 a, m | |
| 16 | 40.1, CH2 | 1.15–1.30 a, m | |
| 17 | 42.9, C | ||
| 18 | 48.1, CH | 1.35 a, m | |
| 19 | 48.41, CH | 2.38 dd (14.0, 7.0) | 1′, 13, 18, 20, 21, 22, 29, 30 |
| 20 | 151.1, C | ||
| 21 | 68.4, CH | 3.99, m | |
| 22a | 35.7, CH2 | 1.42 a, m | |
| 22b | 1.32 a, m | ||
| 23 | 28.2, CH3 | 1.03, s | |
| 24 | 18.3, CH3 | 0.78, s | |
| 25 | 16.1, CH3 | 0.84, s | |
| 26 | 16.3, CH3 | 0.85, s | |
| 27 | 14.3, CH3 | 0.94, s | |
| 28 | 16.9, CH3 | 0.87, s | |
| 29a | 109.5, CH2 | 4.69, s | |
| 29b | 4.57, s | ||
| 30 | 18.1, CH3 | 1.68, s | |
| 1′ | 173.0, C | ||
| 2′ | 36.7, CH2 | 2.45, t (7) | |
| 3′ | 25.2, CH2 | 1.62, m | |
| 4′ | 29.5, CH2 | 1.18–1.28 a, m | |
| 5′ | 29.6, CH2 | 1.18–1.28 a, m | |
| 6′ | 29.7, CH2 | 1.18–1.28 a, m | |
| 7′ | 29.9, CH2 | 1.18–1.28 a, m | |
| 8′ | 29.8, CH2 | 1.18–1.28 a m | |
| 9′ | 29.8, CH2 | 1.18–1.28 a, m | |
| 10′ | 29.8, CH2 | 1.18–1.28 a, m | |
| 11′ | 29.7, CH2 | 1.18–1.28 a, m | |
| 12′ | 32.1, CH2 | 1.32 a, m | |
| 13′ | 22.8, CH2 | 0.79 | |
| 14′ | 14.7, CH3 | 0.88, t (7.0) | |
| OH | 2.94, br |
| Position | δC (ppm) | δH (ppm) (J in Hz) | HMBC (1H → 13C) |
|---|---|---|---|
| 1 | 130.7, C | ||
| 2 | 114.7, CH | 6.69, (br s) | |
| 3 | 144.7, C | ||
| 4 | 147.0, C | ||
| 5 | 113.1, CH | 6.63, d (7.9) | |
| 6 | 123.6, CH | 6.84, d (7.9) | |
| 7 | 76.9, C | ||
| 8a | 42.4, CH2 | 3.10, d (13.7) | 1, 2, 6, 7, 8′, 9′ |
| 8b | 2.91, d (13.7) | 1, 2, 6, 7, 8′, 9′ | |
| 9 | 179.1, C | ||
| 1′ | 126.5, C | ||
| 2′ | 111.9, CH | 6.60, (br s) | |
| 3′ | 147.0, C | ||
| 4′ | 145.4, C | ||
| 5′ | 114.9, CH | 6.82, d (7.9) | |
| 6′ | 121.9, CH | 6.62, d (7.9) | |
| 7′ a | 31.9, CH2 | 2.59, dd (9.4, 4.6) | 1′, 2′, 6′, 8′, 9′, 7, 8 |
| 7′ b | 2.49, dd (9.1, 4.5) | 1′, 2′, 6′, 8′, 9′, 7, 8 | |
| 8′ | 44.2, CH | 2.52, m | 1, 7, 8, 9, 2′, 6′, 7′, 9′ |
| 9′ a | 70.7, CH2 | 4.04, dd (8.9, 6.7) | 7, 9, 7′, 8′ |
| 9′ b | 3.99, dd (8.9, 7.8) | 7, 9, 7′, 8′ | |
| 4-OCH3 | 56.4, CH3 | 3.85, s | 3, 4 |
| 4′-OCH3 | 56.3, CH3 | 3.64, s | 3′, 4′ |
| OH | 5.5–5.8, (broad) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Rubaiai, F.; Al-Shariqi, Z.Z.; Al-Shabibi, K.S.; Husband, J.; Al-Hattali, A.M.; Goettert, M.; Laufer, S.; Baqi, Y.; Hassan, S.I.; Fatope, M.O. Isolation and Identification of Phytocompounds from Maytenus dhofarensis and Their Biological Potentials. Molecules 2023, 28, 6077. https://doi.org/10.3390/molecules28166077
Al-Rubaiai F, Al-Shariqi ZZ, Al-Shabibi KS, Husband J, Al-Hattali AM, Goettert M, Laufer S, Baqi Y, Hassan SI, Fatope MO. Isolation and Identification of Phytocompounds from Maytenus dhofarensis and Their Biological Potentials. Molecules. 2023; 28(16):6077. https://doi.org/10.3390/molecules28166077
Chicago/Turabian StyleAl-Rubaiai, Fatma, Zakiya Zahran Al-Shariqi, Khalsa S. Al-Shabibi, John Husband, Asmaa M. Al-Hattali, Marcia Goettert, Stefan Laufer, Younis Baqi, Syed Imran Hassan, and Majekodunmi O. Fatope. 2023. "Isolation and Identification of Phytocompounds from Maytenus dhofarensis and Their Biological Potentials" Molecules 28, no. 16: 6077. https://doi.org/10.3390/molecules28166077