
Computational Investigation of Conformational Properties of Short Azapeptides: Insights from DFT Study and NBO Analysis
 , ,
, ,
Abstract
:
1. Introduction
2. Results and Discussion
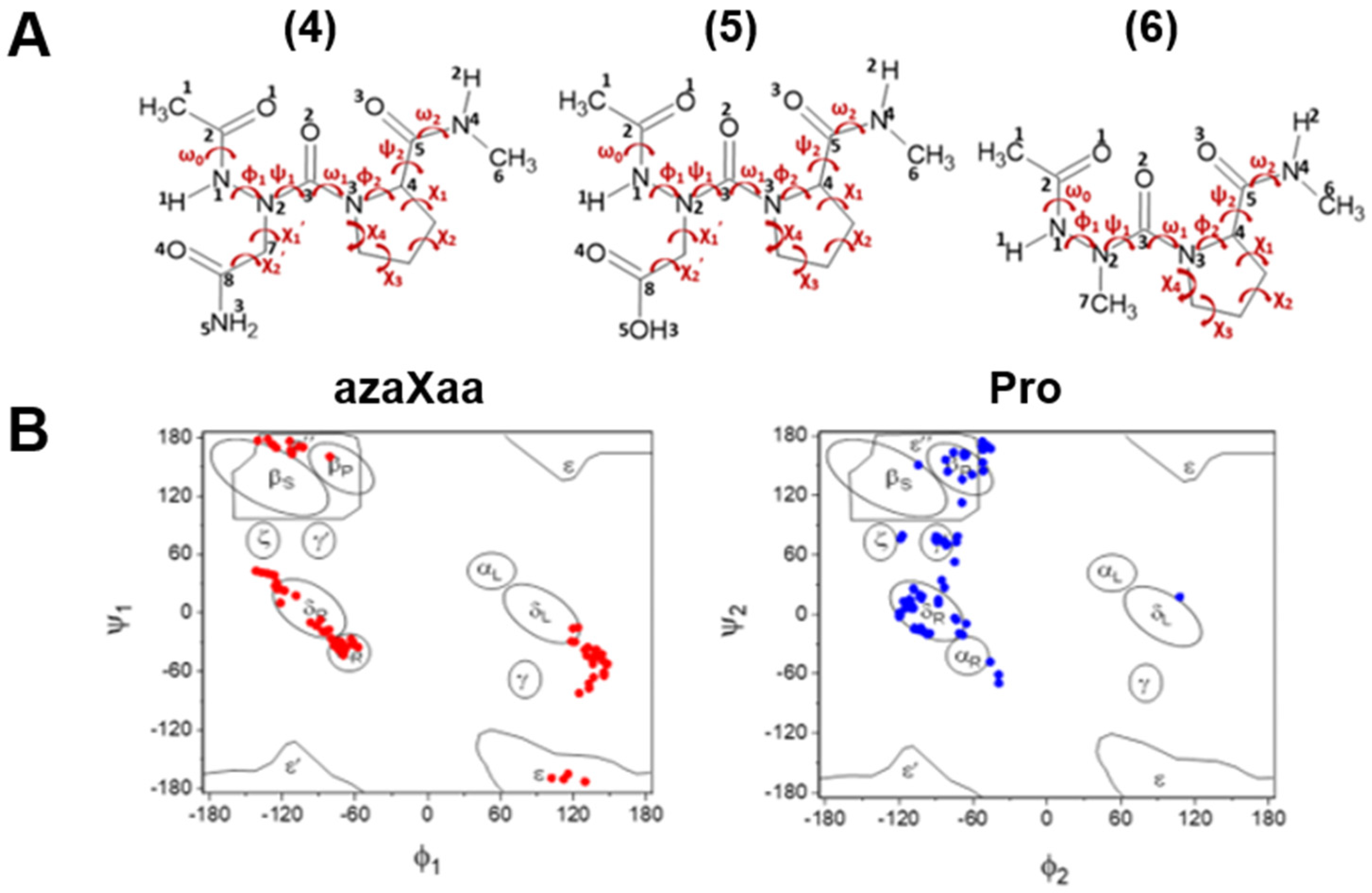
2.1. Cis-Trans Isomerization for Minimum Energy Conformations
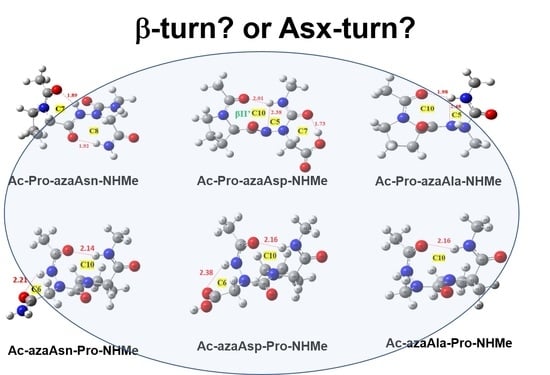
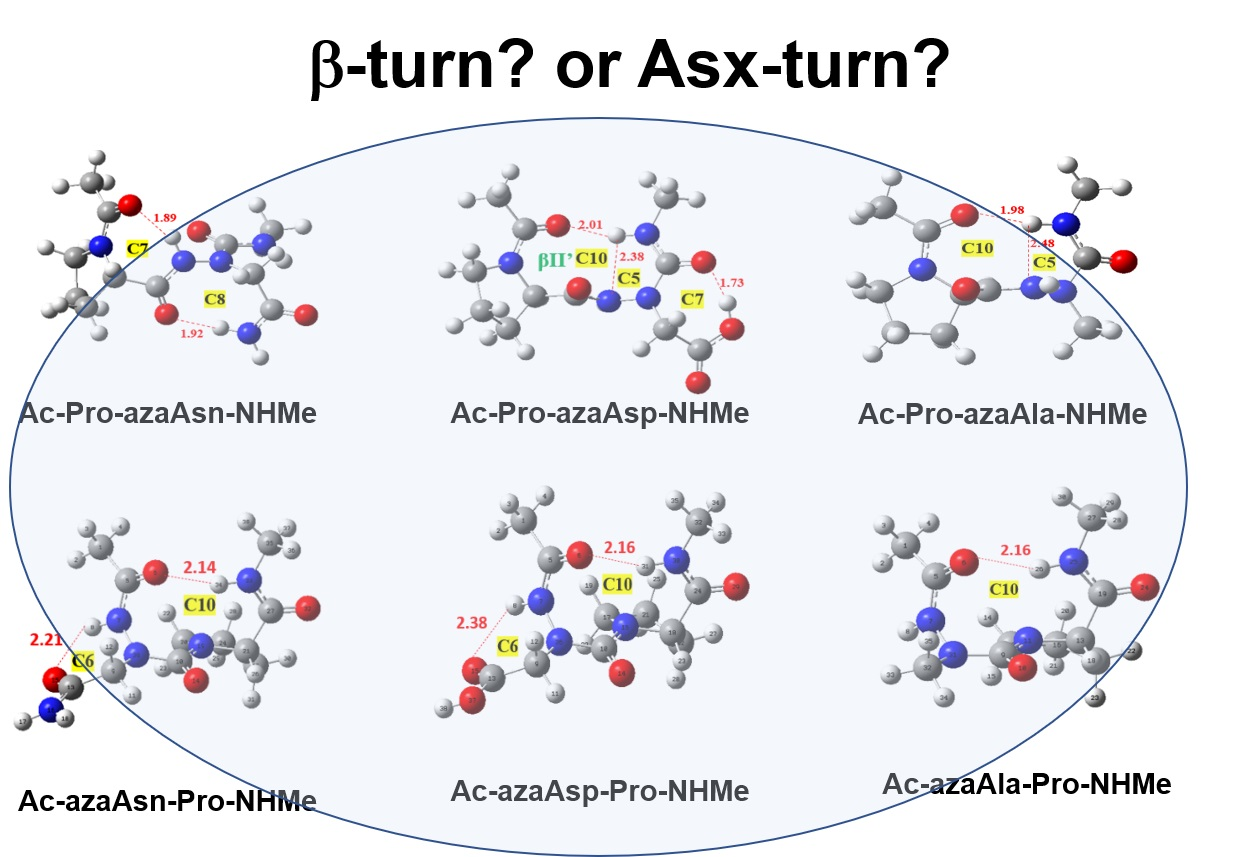
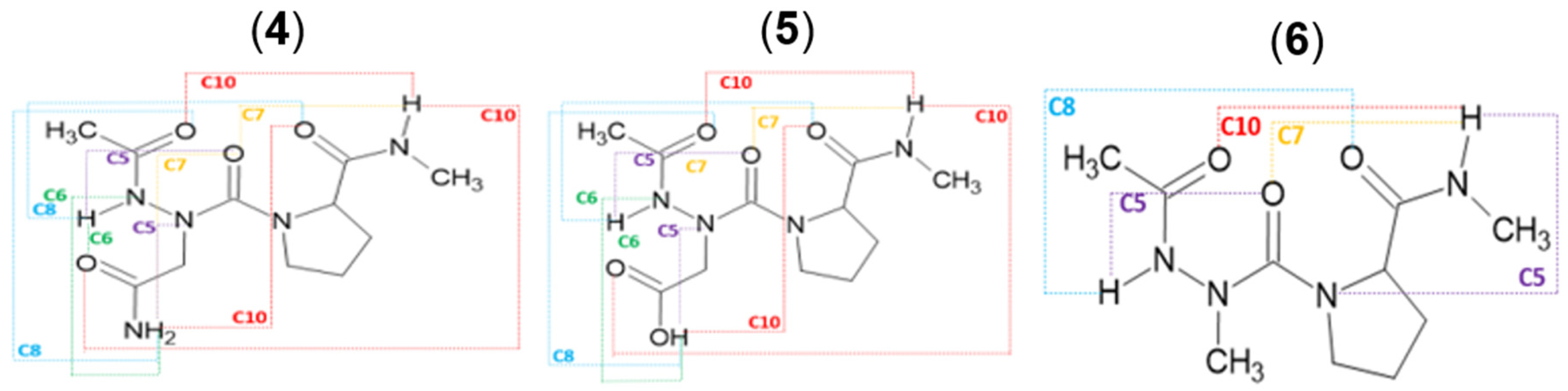
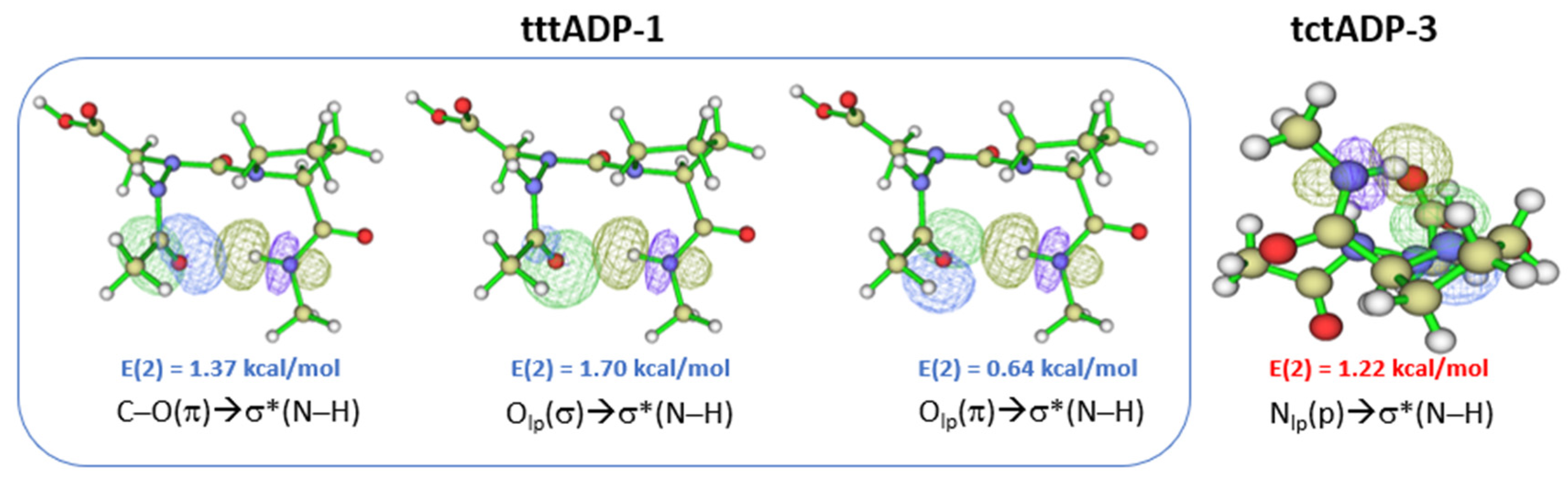
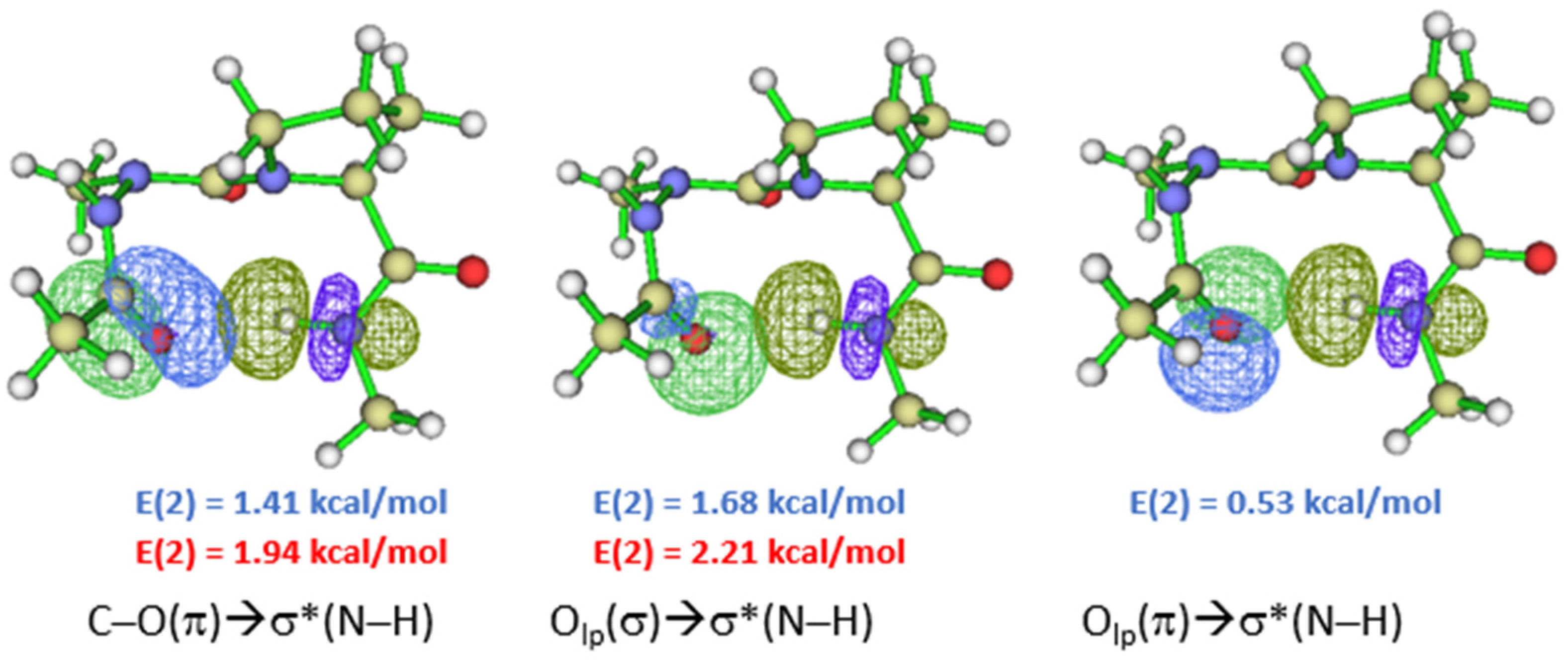
2.2. Hydrogen Bonds in Ac-azaXaa-Pro-NHMe
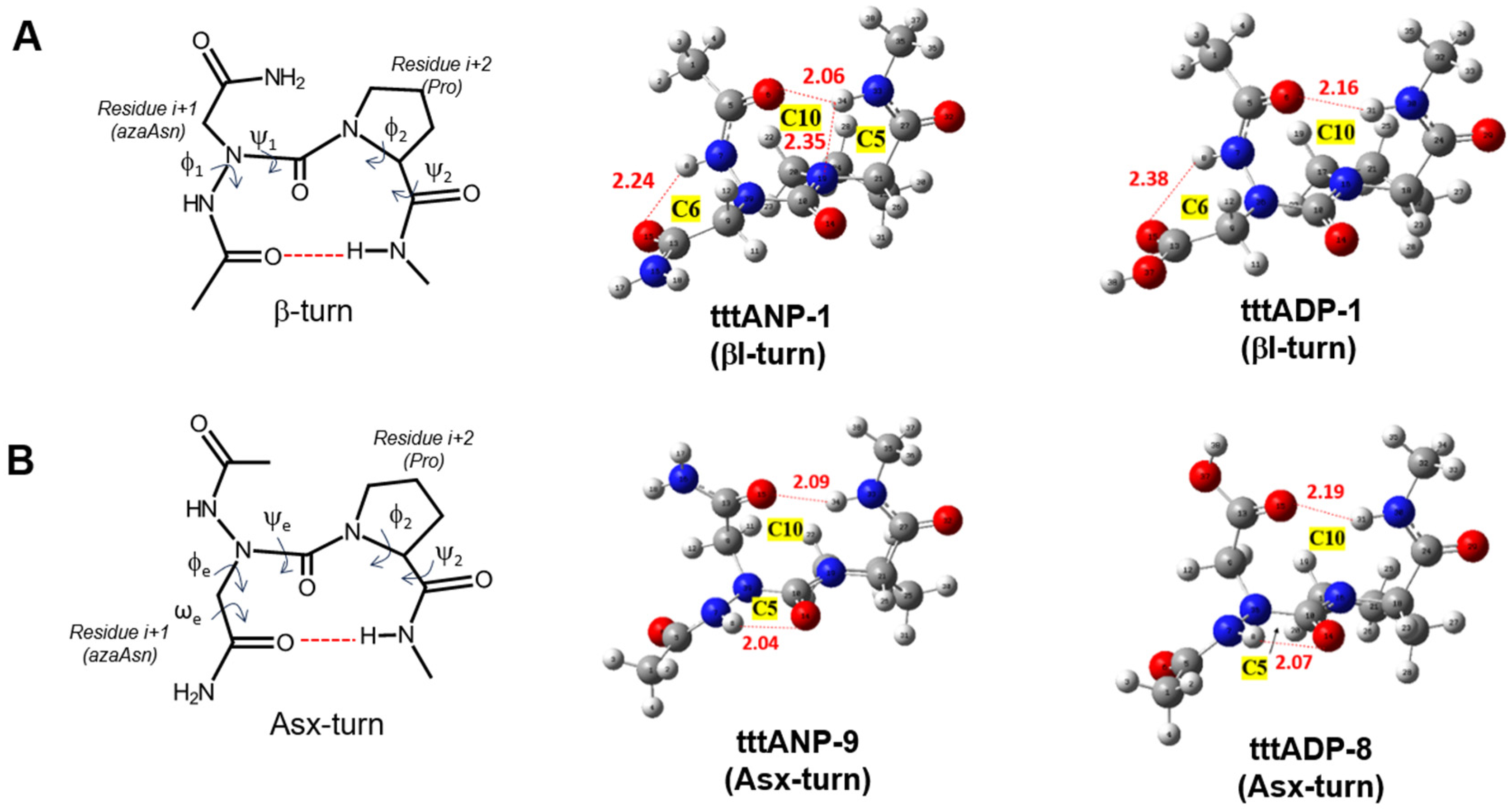
2.3. Asx Turn vs. βⅠ-Tun in Ac-azaAsx-Pro-NHMe (x = n or p)
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Barman, P.; Joshi, S.; Sharma, S.; Preet, S.; Sharma, S.; Saini, A. Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics. Int. J. Pept. Res. Ther. 2023, 29, 61. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Wątły, J.; Miller, A.; Kozłowski, H.; Rowińska-Żyrek, M. Peptidomimetics–An infinite reservoir of metal binding motifs in metabolically stable and biologically active molecules. J. Inorg. Biochem. 2021, 217, 111386. [Google Scholar] [CrossRef] [PubMed]
- Avan, I.; Hall, C.D.; Katritzky, A.R. Peptidomimetics via modifications of amino acids and peptide bonds. Chem. Soc. Rev. 2014, 43, 3575–3594. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.F.; VanPatten, S.; He, M.; Al-Abed, Y. Azapeptides -A History of Synthetic Milestones and Key Examples. Curr. Med. Chem. 2022, 29, 6336–6358. [Google Scholar] [CrossRef]
- Zega, A. Azapeptides as pharmacological agents. Curr. Med. Chem. 2005, 12, 589–597. [Google Scholar] [CrossRef]
- Arujõe, M.; Järv, J.; Mastitski, A.; Ploom, A.; Troska, A. Aza-peptides: Expectations and reality. Proc. Est. Acad. Sci. 2022, 71, 241–254. [Google Scholar] [CrossRef]
- Härk, H.H.; Troska, A.; Arujõe, M.; Burk, P.; Järv, J.; Ploom, A. Kinetic study of aza-amino acid incorporation into peptide chains: Influence of the steric effect of the side chain. Tetrahedron 2022, 129, 133161. [Google Scholar] [CrossRef]
- Proulx, C.; Zhang, J.; Sabatino, D.; Chemtob, S.; Ong, H.; Lubell, W.D. Synthesis and Biomedical Potential of Azapeptide Modulators of the Cluster of Differentiation 36 Receptor (CD36). Biomedicines 2020, 8, 241. [Google Scholar] [CrossRef] [PubMed]
- Bourguet, C.B.; Boulay, P.L.; Claing, A.; Lubell, W.D. Design and synthesis of novel azapeptide activators of apoptosis mediated by caspase-9 in cancer cells. Bioorg. Med. Chem. Lett. 2014, 24, 3361–3365. [Google Scholar] [CrossRef]
- Mhidia, R.; Melnyk, O. Selective cleavage of an azaGly peptide bond by copper(II). Long-range effect of histidine residue. J. Pept. Sci. 2010, 16, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, C.; Desmoulins, P.O.; Driancourt, M.A.; Goericke-Pesch, S.; Hoffmann, B. Reversible downregulation of endocrine and germinative testicular function (hormonal castration) in the dog with the GnRH-agonist azagly-nafarelin as a removable implant “Gonazon”; a preclinical trial. Theriogenology 2009, 71, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.L.; Nestor, J.J., Jr.; McCrae, G.I.; Vickery, B.H. Hydrophobic, aza-glycine analogues of luteinizing hormone-releasing hormone. Int. J. Pept. Protein Res. 1984, 24, 79–84. [Google Scholar] [CrossRef]
- Dutta, A.S.; Furr, B.J.; Giles, M.B.; Valcaccia, B.; Walpole, A.L. Potent agonist and antagonist analogues of luliberin containing an azaglycine residue in position 10. Biochem. Biophys. Res. Commun. 1978, 81, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Kim, J.H.; Jung, H.J.; Kim, K.-Y.; Kim, E.-J.; Choi, Y.-S.; Yoon, C.-J. Computational study of conformational preferences of thioamide-containing azaglycine peptides. J. Comput. Chem. 2004, 25, 169–178. [Google Scholar] [CrossRef]
- Lee, H.-J.; Jung, H.J.; Kim, J.H.; Park, H.-M.; Lee, K.-B. Conformational preference of azaglycine-containing dipeptides studied by PCM and IPCM methods. Chem. Phys. 2003, 294, 201–210. [Google Scholar] [CrossRef]
- Lee, H.; Song, J.; Choi, Y.; Ro, S.; Yoon, C. The energetically favorable cis peptide bond for the azaglycine-containing peptide: For-AzGly-NH2 model. Phys. Chem. Chem. Phys. 2001, 3, 1693–1698. [Google Scholar] [CrossRef]
- Kang, Y.K.; Byun, B.J. Conformational preferences and cis-trans isomerization of azaproline residue. J. Phys. Chem. B 2007, 111, 5377–5385. [Google Scholar] [CrossRef]
- Lee, H.J.; Song, J.W.; Choi, Y.S.; Park, H.M.; Lee, K.B. A theoretical study of conformational properties of N-methyl azapeptide derivatives. J. Am. Chem. Soc. 2002, 124, 11881–11893. [Google Scholar] [CrossRef]
- Lee, H.J.; Park, H.M.; Lee, K.B. The beta-turn scaffold of tripeptide containing an azaphenylalanine residue. Biophys. Chem. 2007, 125, 117–126. [Google Scholar] [CrossRef]
- El Khabchi, M.; Lahlou, H.; El Adnani, Z.; McHarfi, M.; Benzakour, M.; Fitri, A.; Benjelloun, A.T. Conformational preferences of Ac-Pro-azaXaa-NHMe (Xaa = Asn, Asp, Ala) and the effect of intramolecular hydrogen bonds on their stability in gas phase and solution. J. Mol. Model 2021, 27, 368. [Google Scholar] [CrossRef] [PubMed]
- Bowles, M.; Proulx, C. Solid phase submonomer azapeptide synthesis. Methods Enzym. 2021, 656, 169–190. [Google Scholar] [CrossRef]
- Dai, C.; Ma, J.; Li, M.; Wu, W.; Xia, X.; Zhang, J. Diversity-oriented submonomer synthesis of azapeptides mediated by the Mitsunobu reaction. Org. Chem. Front. 2019, 6, 2529–2533. [Google Scholar] [CrossRef]
- Chingle, R.M.; Proulx, C.; Lubell, W.D. Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. Acc. Chem. Res. 2017, 50, 1541–1556. [Google Scholar] [CrossRef]
- Doan, N.D.; Zhang, J.; Traoré, M.; Kamdem, W.; Lubell, W.D. Solid-phase synthesis of C-terminal azapeptides. J. Pept. Sci. 2015, 21, 387–391. [Google Scholar] [CrossRef]
- Ollivier, N.; Besret, S.; Blanpain, A.; Melnyk, O. Silver-catalyzed azaGly ligation. Application to the synthesis of azapeptides and of lipid-peptide conjugates. Bioconjug. Chem. 2009, 20, 1397–1403. [Google Scholar] [CrossRef]
- Bowles, M.; Proulx, C. Late-Stage N-Alkylation of Azapeptides. Org. Lett. 2022, 24, 1768–1773. [Google Scholar] [CrossRef]
- Luo, Z.; Xu, L.; Tang, X.; Zhao, X.; He, T.; Lubell, W.D.; Zhang, J. Synthesis and biological evaluation of novel all-hydrocarbon cross-linked aza-stapled peptides. Org. Biomol. Chem. 2022, 20, 7963–7971. [Google Scholar] [CrossRef]
- Singh, S.; Shrivastava, R.; Singh, G.; Ali, R.; Sankar Ampapathi, R.; Bhadhuria, S.; Haq, W. AzaGly-Appended Peptidomimetics Structurally Related to PTR6154 as Potential PKB/Akt Inhibitors. Chembiochem 2017, 18, 1061–1065. [Google Scholar] [CrossRef]
- Sabatino, D.; Proulx, C.; Pohankova, P.; Ong, H.; Lubell, W.D. Structure-activity relationships of GHRP-6 azapeptide ligands of the CD36 scavenger receptor by solid-phase submonomer azapeptide synthesis. J. Am. Chem. Soc. 2011, 133, 12493–12506. [Google Scholar] [CrossRef]
- Altiti, A.S.; He, M.; VanPatten, S.; Cheng, K.F.; Ahmed, U.; Chiu, P.Y.; Mughrabi, I.T.; Jabari, B.A.; Burch, R.M.; Manogue, K.R.; et al. Thiocarbazate building blocks enable the construction of azapeptides for rapid development of therapeutic candidates. Nat. Commun. 2022, 13, 7127. [Google Scholar] [CrossRef]
- Karplus, P.A. Experimentally observed conformation-dependent geometry and hidden strain in proteins. Protein Sci. 1996, 5, 1406–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melton, S.D.; Smith, M.S.; Chenoweth, D.M. Incorporation of Aza-Glycine into Collagen Peptides. J. Org. Chem. 2020, 85, 1706–1711. [Google Scholar] [CrossRef]
- Etzkorn, F.A.; Ware, R.I.; Pester, A.M.; Troya, D. Conformational Analysis of n→π* Interactions in Collagen Triple Helix Models. J. Phys. Chem. B 2019, 123, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Melton, S.D.; Chenoweth, D.M. Variation in the Yaa position of collagen peptides containing azaGlycine. Chem. Commun. 2018, 54, 11937–11940. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Malamakal, R.M.; Chenoweth, D.M. Aza-Glycine Induces Collagen Hyperstability. J. Am. Chem. Soc. 2015, 137, 12422–12425. [Google Scholar] [CrossRef]
- Harris, T.; Chenoweth, D.M. Sterics and Stereoelectronics in Aza-Glycine: Impact of Aza-Glycine Preorganization in Triple Helical Collagen. J. Am. Chem. Soc. 2019, 141, 18021–18029. [Google Scholar] [CrossRef]
- Kasznel, A.J.; Harris, T.; Porter, N.J.; Zhang, Y.; Chenoweth, D.M. Aza-proline effectively mimics l-proline stereochemistry in triple helical collagen. Chem. Sci. 2019, 10, 6979–6983. [Google Scholar] [CrossRef] [Green Version]
- Kasznel, A.J.; Zhang, Y.; Hai, Y.; Chenoweth, D.M. Structural Basis for Aza-Glycine Stabilization of Collagen. J. Am. Chem. Soc. 2017, 139, 9427–9430. [Google Scholar] [CrossRef]
- Ro, S.; Lee, H.J.; Ahn, I.; Shin, D.; Lee, K.; Yoon, C.; Choi, Y. Torsion angle based design of peptidomimetics: A dipeptidic template adopting beta-I turn (Ac-Aib-AzGly-NH2). Bioorg. Med. Chem. 2001, 9, 1837–1841. [Google Scholar] [CrossRef]
- McMechen, M.A.; Willis, P.C.; Proulx, G.C. Aza-amino acids disrupt b-sheet secondary structures. Molecules 2019, 24, 1919. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Brenner, V.; Gloaguen, E.; Mons, M. Rationalizing the diversity of amide–amide H-bonding in peptides using the natural bond orbital method. Phys. Chem. Chem. Phys. 2019, 21, 24601–24619. [Google Scholar] [CrossRef] [PubMed]
- Alemán, C.; Puiggalí, J. Conformational Preferences of the Asparagine Residue. Gas-Phase, Aqueous Solution, and Chloroform Solution Calculations on the Model Dipeptide. J. Phys. Chem. B 1997, 101, 3441–3446. [Google Scholar] [CrossRef]
- André, F.; Vicherat, A.; Boussard, G.; Aubry, A.; Marraud, M. Aza-peptides. III. Experimental structural analysis of aza-alanine and aza-asparagine-containing peptides. J. Pept. Res. 1997, 50, 372–381. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Boussard, G.; Bayeul, D.; Didierjean, C.; Aubry, A.; Marraud, M. Aza-peptides. II. X-ray structures of aza-alanine and aza-asparagine-containing peptides. J. Pept. Res. 1997, 49, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Baruah, K.; Sahariah, B.; Sakpal, S.S.; Deka, J.K.R.; Bar, A.K.; Bagchi, S.; Sarma, B.K. Stabilization of Azapeptides by N(amide)···H-N(amide) Hydrogen Bonds. Org. Lett. 2021, 23, 4949–4954. [Google Scholar] [CrossRef]
- Abbadi, A.; McHarfi, M.; Aubry, A.; Premilat, S.; Boussard, G.; Marraud, M. Involvement of side functions in peptide structures: The Asx turn. Occurrence and conformational aspects. J. Am. Chem. Soc. 1991, 113, 2729–2735. [Google Scholar] [CrossRef]
- Duddy, W.J.; Nissink, J.W.; Allen, F.H.; Milner-White, E.J. Mimicry by asx- and ST-turns of the four main types of β-turn in proteins. Protein Sci. 2004, 13, 3051–3055. [Google Scholar] [CrossRef] [Green Version]
- D’Mello, V.C.; Goldsztejn, G.; Rao Mundlapati, V.; Brenner, V.; Gloaguen, E.; Charnay-Pouget, F.; Aitken, D.J.; Mons, M. Characterization of Asx Turn Types and Their Connate Relationship with β-Turns. Chemistry 2022, 28, e202104328. [Google Scholar] [CrossRef] [PubMed]
- Froimowitz, M. HyperChem: A software package for computational chemistry and molecular modeling. Biotechniques 1993, 14, 1010–1013. [Google Scholar] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Software: Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ac-azaAsn-Pro-NHMe | Ac-azaAsp-Pro-NHMe | Ac-azaAla-Pro-NHMe | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔE(M1) | ΔE(M2) | ΔE(M1) | ΔE(M2) | ΔE(M1) | ΔE(M2) | |||
| tttANP-1 | a 0.00 | 0.94 | tttADP-1 | c 0.00 | 0.69 | tttAAP-1 | e 0.00 | f 0.00 |
| tctANP-2 | 0.94 | b 0.00 | tttADP-2 | 1.00 | 2.70 | tttAAP-2 | 0.75 | 1.13 |
| tttANP-3 | 1.32 | 2.64 | tctADP-3 | 1.07 | d 0.00 | tttAAP-3 | 1.00 | 2.51 |
| tttANP-4 | 1.38 | 1.95 | tctADP-4 | 1.44 | 0.38 | tttAAP-4 | 1.26 | 3.70 |
| tttANP-5 | 1.96 | 2.89 | cttADP-5 | 2.07 | 2.57 | tctAAP-5 | 1.44 | 1.82 |
| cttANP-6 | 2.45 | 3.07 | tttADP-6 | 2.95 | 3.33 | cttAAP-6 | 1.51 | 2.26 |
| tctANP-7 | 2.64 | 2.01 | tttADP-7 | 3.33 | 4.39 | cttAAP-7 | 1.63 | 2.20 |
| tttANP-8 | 2.64 | 3.20 | tttADP-8 | 3.70 | 5.46 | tctAAP-8 | 2.26 | 1.26 |
| tttANP-9 | 2.89 | 4.52 | tttADP-9 | 4.52 | 5.33 | tttAAP-9 | 2.70 | 4.89 |
| cttANP-10 | 3.07 | 3.26 | cttADP-10 | 4.52 | 4.58 | tctAAP-10 | 2.76 | 2.20 |
| cttANP-11 | 3.26 | 2.95 | cttADP-11 | 4.58 | 4.83 | cttAAP-11 | 3.51 | 3.01 |
| tttANP-12 | 4.08 | 4.89 | cctADP-12 | 4.83 | 3.39 | tctAAP-12 | 3.70 | 5.27 |
| tctANP-13 | 4.27 | 3.20 | tctADP-13 | 5.08 | 4.89 | cctAAP-13 | 3.77 | 2.01 |
| tctANP-14 | 4.46 | 3.33 | tttADP-14 | 5.65 | 7.53 | tttAAP-14 | 3.89 | 4.64 |
| tttANP-15 | 4.46 | 5.65 | cctADP-15 | 6.02 | 4.89 | cctAAP-15 | 4.33 | 2.95 |
| cctANP-16 | 4.52 | 2.32 | tctADP-16 | 6.34 | 3.83 | cttAAP-16 | 4.96 | 6.46 |
| cctANP-17 | 4.64 | 3.20 | tctADP-17 | 6.78 | 7.22 | cctAAP-17 | 5.52 | 5.02 |
| cttANP-18 | 4.83 | 4.96 | cctADP-18 | 6.97 | 4.96 | ttcAAP-18 | 6.28 | 5.58 |
| tctANP-19 | 5.02 | 3.26 | cctADP-19 | 7.40 | 5.40 | cctAAP-19 | 6.34 | 5.46 |
| cttANP-20 | 5.40 | 5.71 | tctADP-20 | 7.66 | 6.21 | cccAAP-20 | 8.47 | 6.15 |
| cctANP-21 | 5.46 | 4.46 | tttADP-21 | 7.72 | 9.04 | tccAAP-21 | 10.10 | 7.66 |
| cctANP-22 | 5.58 | 3.26 | tctADP-22 | 8.03 | 6.97 | cccAAP-22 | 11.92 | 9.73 |
| cttANP-23 | 5.96 | 5.77 | cttADP-23 | 8.03 | 7.97 | |||
| tctANP-24 | 6.71 | 7.22 | ttcADP-24 | 9.29 | 8.84 | |||
| tctANP-25 | 6.90 | 5.52 | tccADP-25 | 10.10 | 7.47 | |||
| cttANP-26 | 7.59 | 7.53 | cttADP-26 | 10.86 | 11.11 | |||
| tttANP-27 | 7.59 | 9.66 | cttADP-27 | 11.30 | 10.73 | |||
| ttcANP-28 | 7.97 | 7.59 | cccADP-28 | 13.43 | 9.22 | |||
| tccANP-29 | 9.16 | 6.21 | cctADP-29 | 13.43 | 12.61 | |||
| tttANP-30 | 9.79 | 11.36 | cccADP-30 | 16.13 | 12.74 | |||
| cctANP-31 | 9.98 | 9.48 | ||||||
| cccANP-32 | 10.35 | 6.34 | ||||||
| cccANP-33 | 14.75 | 11.80 | ||||||
| Ac-azaAsn-Pro-NHMe | Ac-azaAsp-Pro-NHMe | Ac-azaAla-Pro-NHMe | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔE(M3) | ΔE(M4) | ΔE(M3) | ΔE(M4) | ΔE(M3) | ΔE(M4) | |||
| tttANP-1 | 1.13 | 1.51 | tttADP-1 | 3.14 | 2.70 | tttAAP-1 | e 0.00 | f 0.00 |
| tctANP-2 | 1.38 | b 0.00 | tttADP-2 | 5.21 | 5.71 | tttAAP-2 | 2.01 | 3.07 |
| tttANP-3 | 3.07 | 4.52 | tctADP-3 | 3.64 | 1.38 | tttAAP-3 | 2.76 | 4.27 |
| tttANP-4 | 3.01 | 3.07 | tctADP-4 | 5.15 | 3.58 | tttAAP-4 | 2.70 | 6.46 |
| tttANP-5 | 2.76 | 3.45 | cttADP-5 | 6.84 | 6.28 | tctAAP-5 | 3.26 | 3.95 |
| cttANP-6 | 4.64 | 4.96 | tttADP-6 | 4.27 | 3.45 | cttAAP-6 | 4.96 | 5.46 |
| tctANP-7 | 4.27 | 3.70 | tttADP-7 | 2.82 | 2.45 | cttAAP-7 | 5.15 | 5.71 |
| tttANP-8 | 1.51 | 1.57 | tttADP-8 | 5.40 | 5.77 | tctAAP-8 | 2.01 | 0.88 |
| tttANP-9 | 2.95 | 4.20 | tttADP-9 | c 0.00 | d 0.00 | tttAAP-9 | 2.64 | 4.64 |
| cttANP-10 | 6.78 | 6.59 | cttADP-10 | 5.65 | 5.27 | tctAAP-10 | 1.82 | 2.26 |
| cttANP-11 | 7.15 | 6.34 | cttADP-11 | 7.66 | 6.90 | cttAAP-11 | 2.82 | 2.32 |
| tttANP-12 | a 0.00 | 0.82 | cctADP-12 | 6.02 | 3.45 | tctAAP-12 | 5.02 | 6.34 |
| tctANP-13 | 3.33 | 4.89 | tctADP-13 | 5.84 | 4.33 | cctAAP-13 | 4.46 | 3.07 |
| tctANP-14 | 2.95 | 2.07 | tttADP-14 | 6.09 | 6.90 | tttAAP-14 | 1.69 | 6.34 |
| tttANP-15 | 4.64 | 5.84 | cctADP-15 | 9.29 | 7.78 | cctAAP-15 | 4.58 | 3.01 |
| cctANP-16 | 3.64 | 2.38 | tctADP-16 | 6.02 | 5.08 | cttAAP-16 | 3.77 | 4.96 |
| cctANP-17 | 3.77 | 2.07 | tctADP-17 | 4.71 | 4.64 | cctAAP-17 | 5.52 | 5.02 |
| cttANP-18 | 3.51 | 4.02 | cctADP-18 | 7.15 | 4.58 | ttcAAP-18 | 4.33 | 7.22 |
| tctANP-19 | 3.07 | 2.13 | cctADP-19 | 6.02 | 4.14 | cctAAP-19 | 4.46 | 3.95 |
| cttANP-20 | 4.33 | 4.58 | tctADP-20 | 7.40 | 5.15 | cccAAP-20 | 7.34 | 6.21 |
| cctANP-21 | 6.78 | 5.96 | tttADP-21 | 5.08 | 6.84 | tccAAP-21 | 7.91 | 5.58 |
| cctANP-22 | 6.02 | 4.08 | tctADP-22 | 5.15 | 3.45 | cccAAP-22 | 11.04 | 9.29 |
| cttANP-23 | 5.58 | 4.02 | cttADP-23 | 5.90 | 4.83 | |||
| tctANP-24 | 2.70 | 3.51 | ttcADP-24 | 5.15 | 10.42 | |||
| tctANP-25 | 4.14 | 4.20 | tccADP-25 | 10.17 | 6.46 | |||
| cttANP-26 | 3.89 | 3.45 | cttADP-26 | 8.03 | 7.53 | |||
| tttANP-27 | 4.08 | 5.84 | cttADP-27 | 7.59 | 6.21 | |||
| ttcANP-28 | 10.17 | 9.79 | cccADP-28 | 10.73 | 6.40 | |||
| tccANP-29 | 9.35 | 6.15 | cctADP-29 | 9.91 | 8.22 | |||
| tttANP-30 | 3.07 | 5.71 | cccADP-30 | 12.80 | 7.72 | |||
| cctANP-31 | 6.34 | 5.52 | ||||||
| cccANP-32 | 8.22 | 4.64 | ||||||
| cccANP-33 | 12.56 | 10.10 | ||||||
| Turn | ϕi+1 (°) Asx ϕe | ψi+1 (°) Asx ψe | ϕi+2 (°) | ψi+2 (°) |
|---|---|---|---|---|
| βI | −60 | −30 | −90 | 0 |
| βI′ | 60 | 30 | 90 | 0 |
| βII | −60 | 120 | 80 | 0 |
| βII′ | 60 | −120 | −80 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Khabchi, M.; Mcharfi, M.; Benzakour, M.; Fitri, A.; Benjelloun, A.T.; Song, J.-W.; Lee, K.-B.; Lee, H.-J. Computational Investigation of Conformational Properties of Short Azapeptides: Insights from DFT Study and NBO Analysis. Molecules 2023, 28, 5454. https://doi.org/10.3390/molecules28145454
El Khabchi M, Mcharfi M, Benzakour M, Fitri A, Benjelloun AT, Song J-W, Lee K-B, Lee H-J. Computational Investigation of Conformational Properties of Short Azapeptides: Insights from DFT Study and NBO Analysis. Molecules. 2023; 28(14):5454. https://doi.org/10.3390/molecules28145454
Chicago/Turabian StyleEl Khabchi, Mouna, Mohammed Mcharfi, Mohammed Benzakour, Asmae Fitri, Adil Touimi Benjelloun, Jong-Won Song, Kang-Bong Lee, and Ho-Jin Lee. 2023. "Computational Investigation of Conformational Properties of Short Azapeptides: Insights from DFT Study and NBO Analysis" Molecules 28, no. 14: 5454. https://doi.org/10.3390/molecules28145454