
Synthesis and Structural Characterization of Copper Complexes Containing “R-Substituted” Bis-7-Azaindolyl Borate Ligands

 ,
,
Abstract
:
1. Introduction
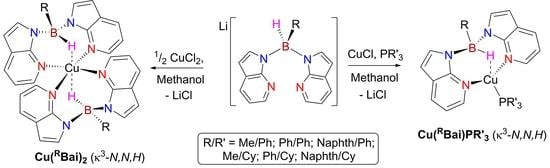
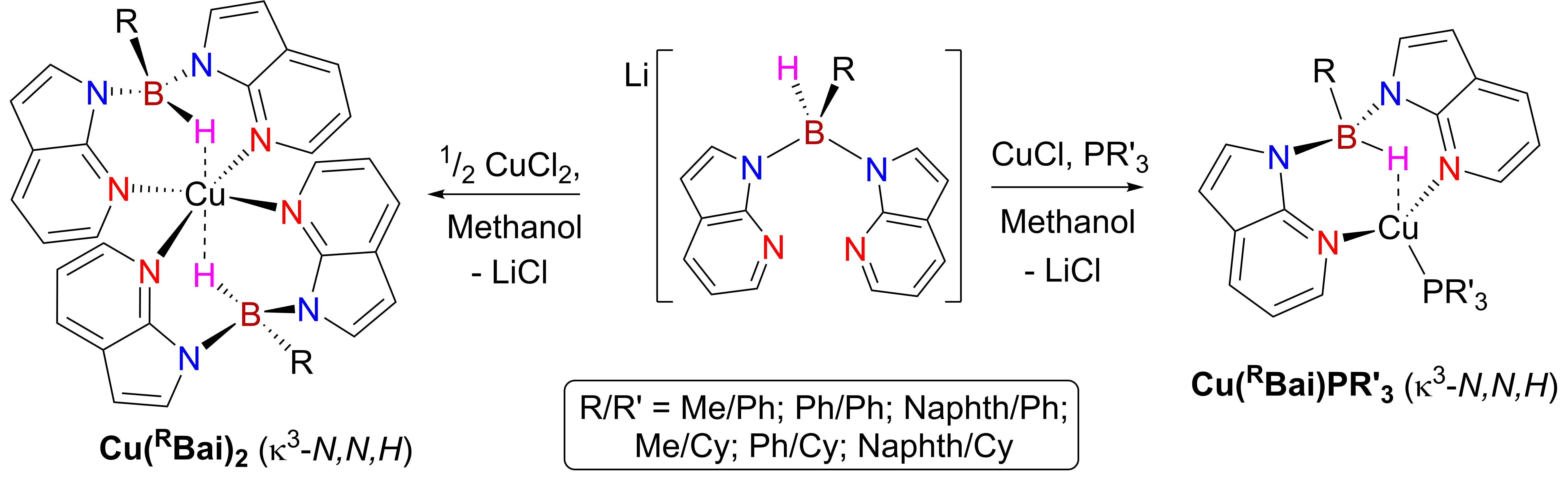
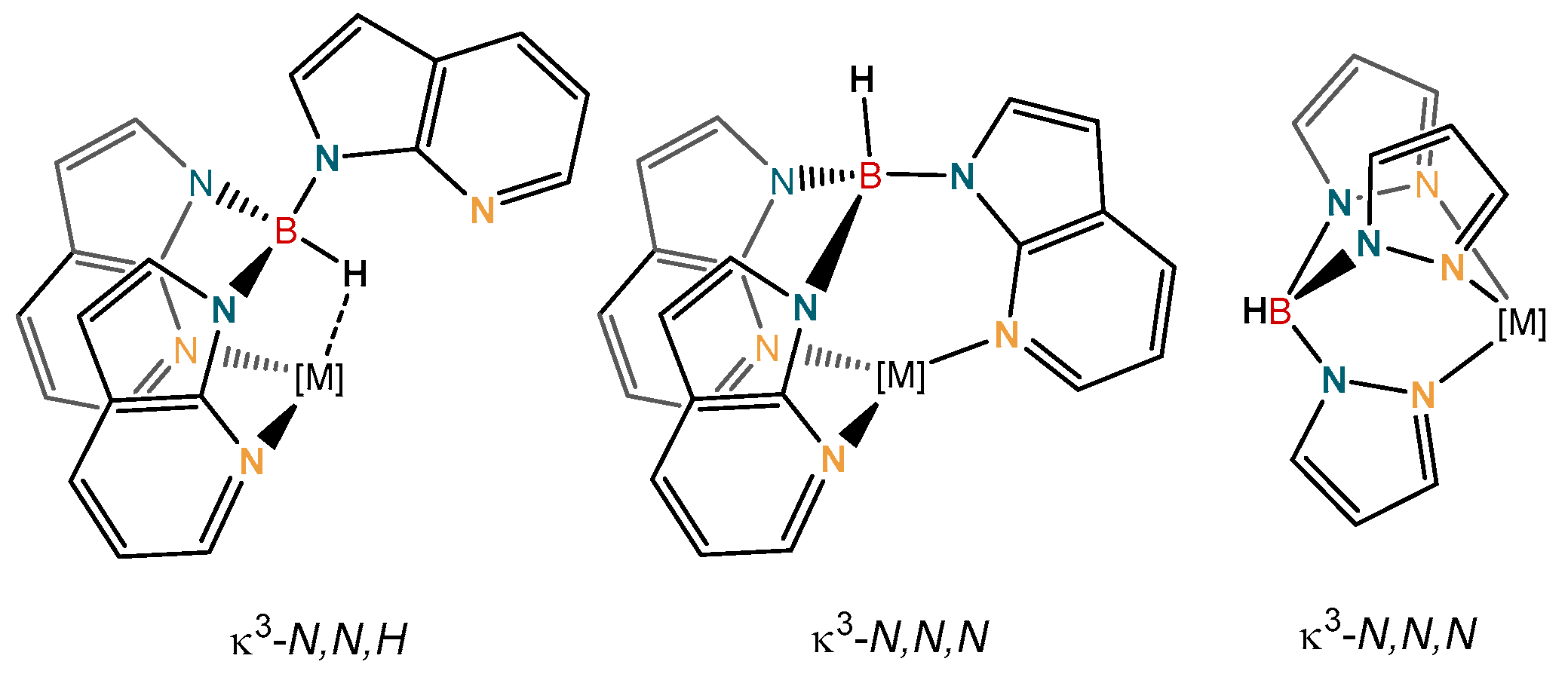
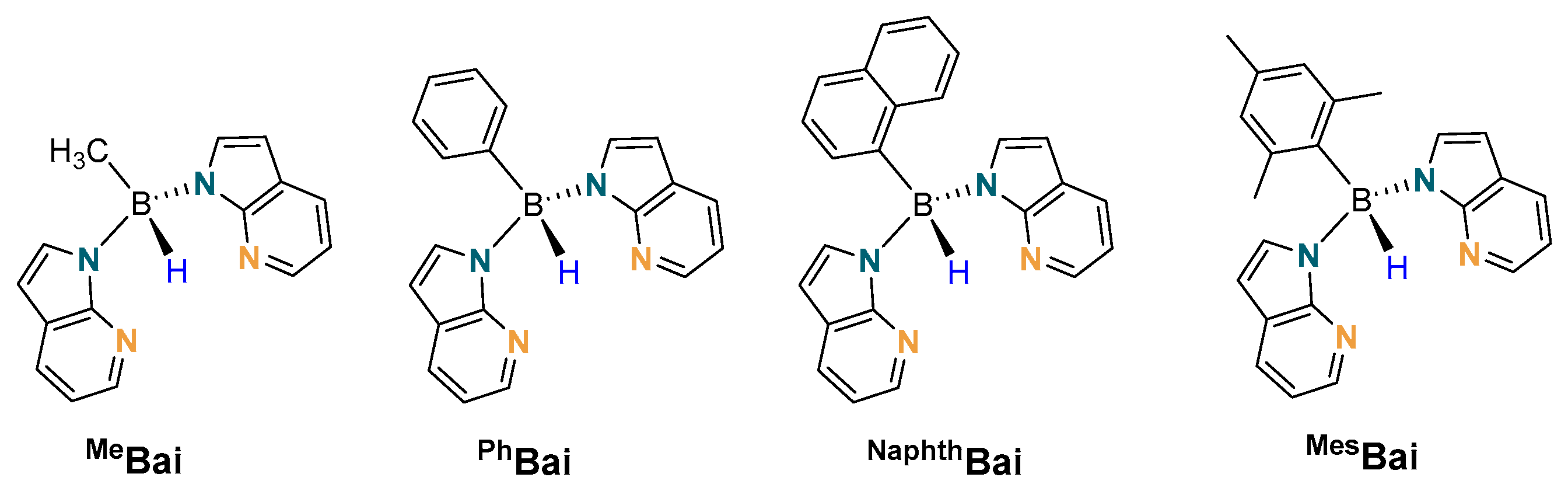
2. Results and Discussion
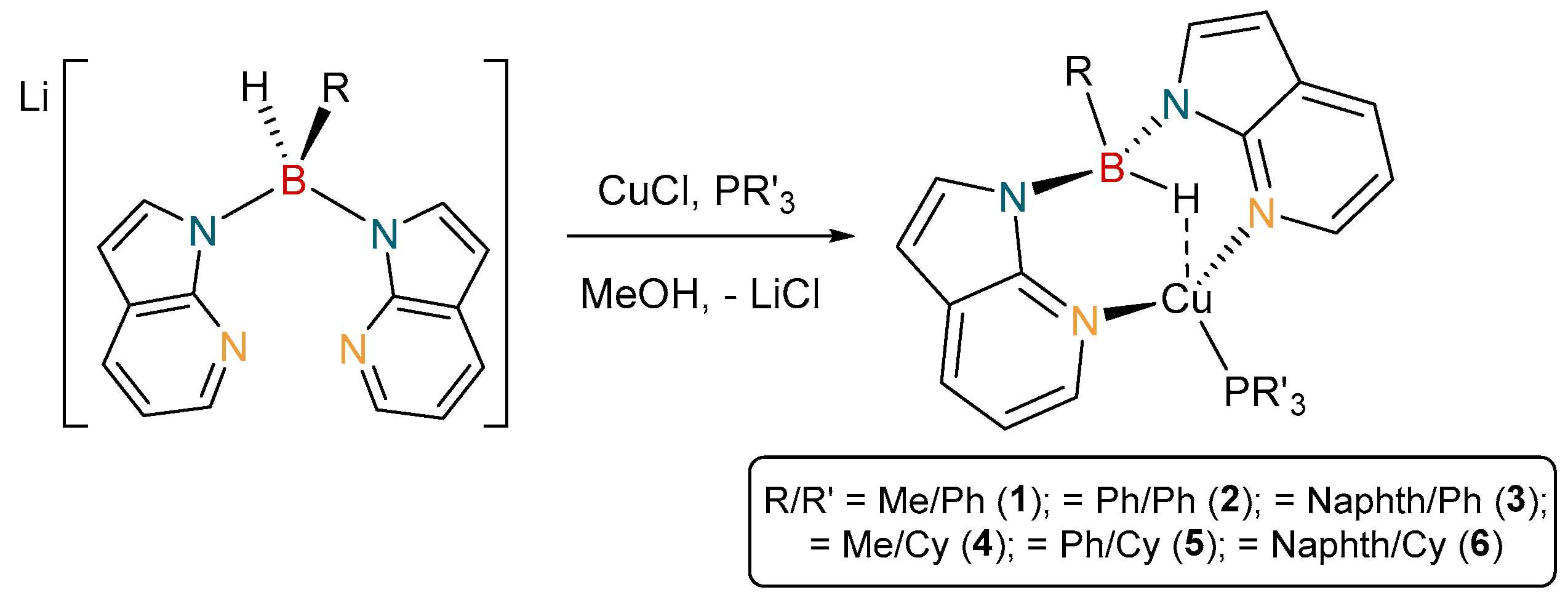
2.1. Synthesis and Characterisation of Copper(I) Phosphine Complexes
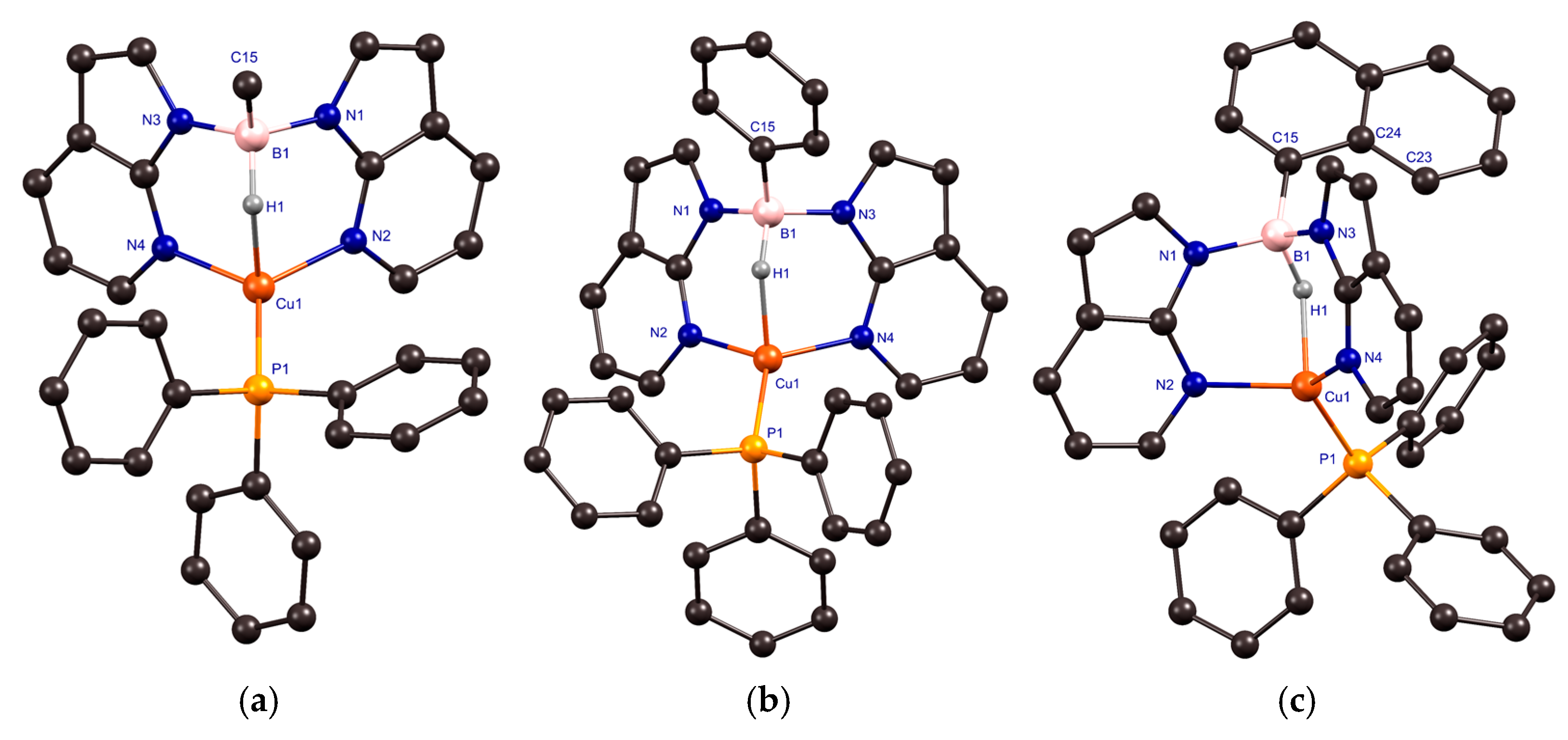
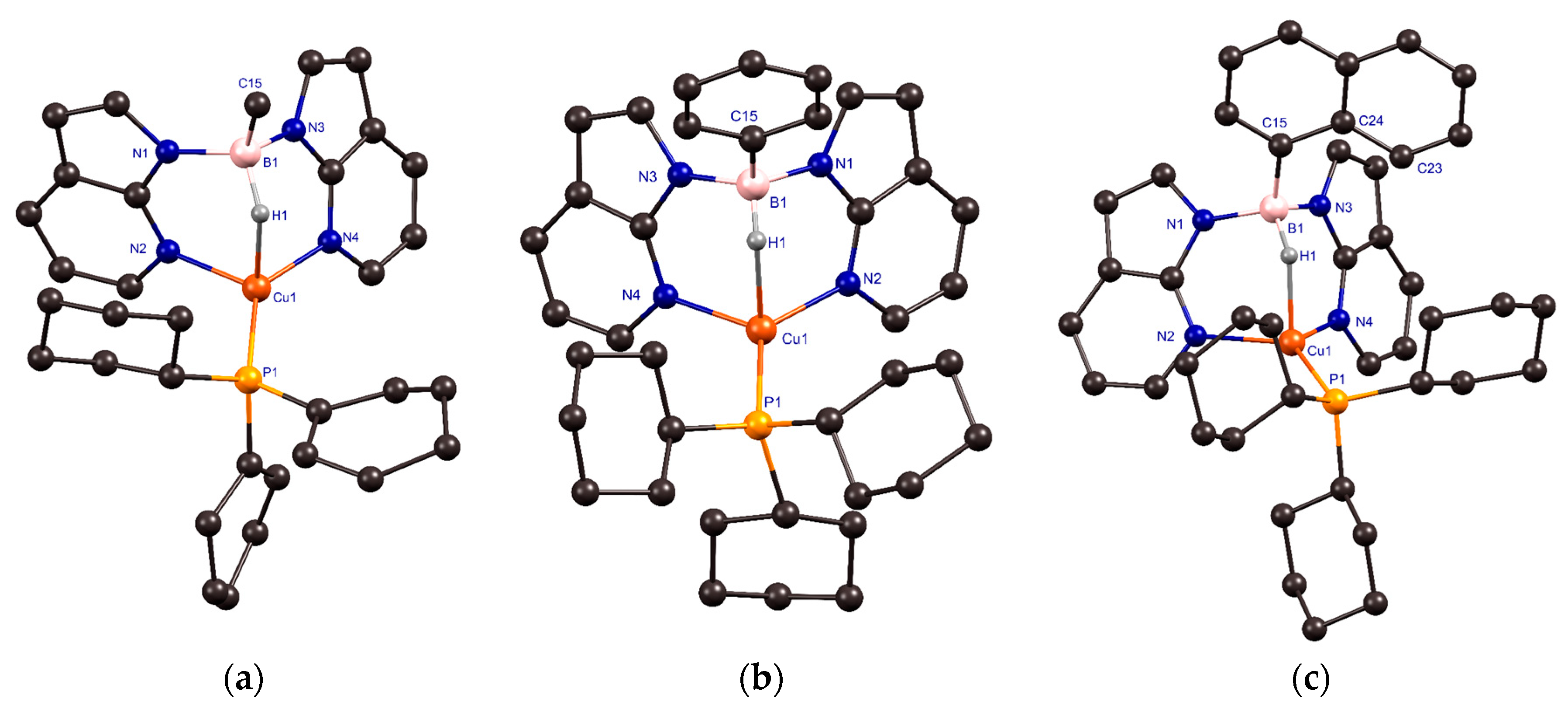
2.2. Structural Characterization of Copper(I) Phosphine Complexes
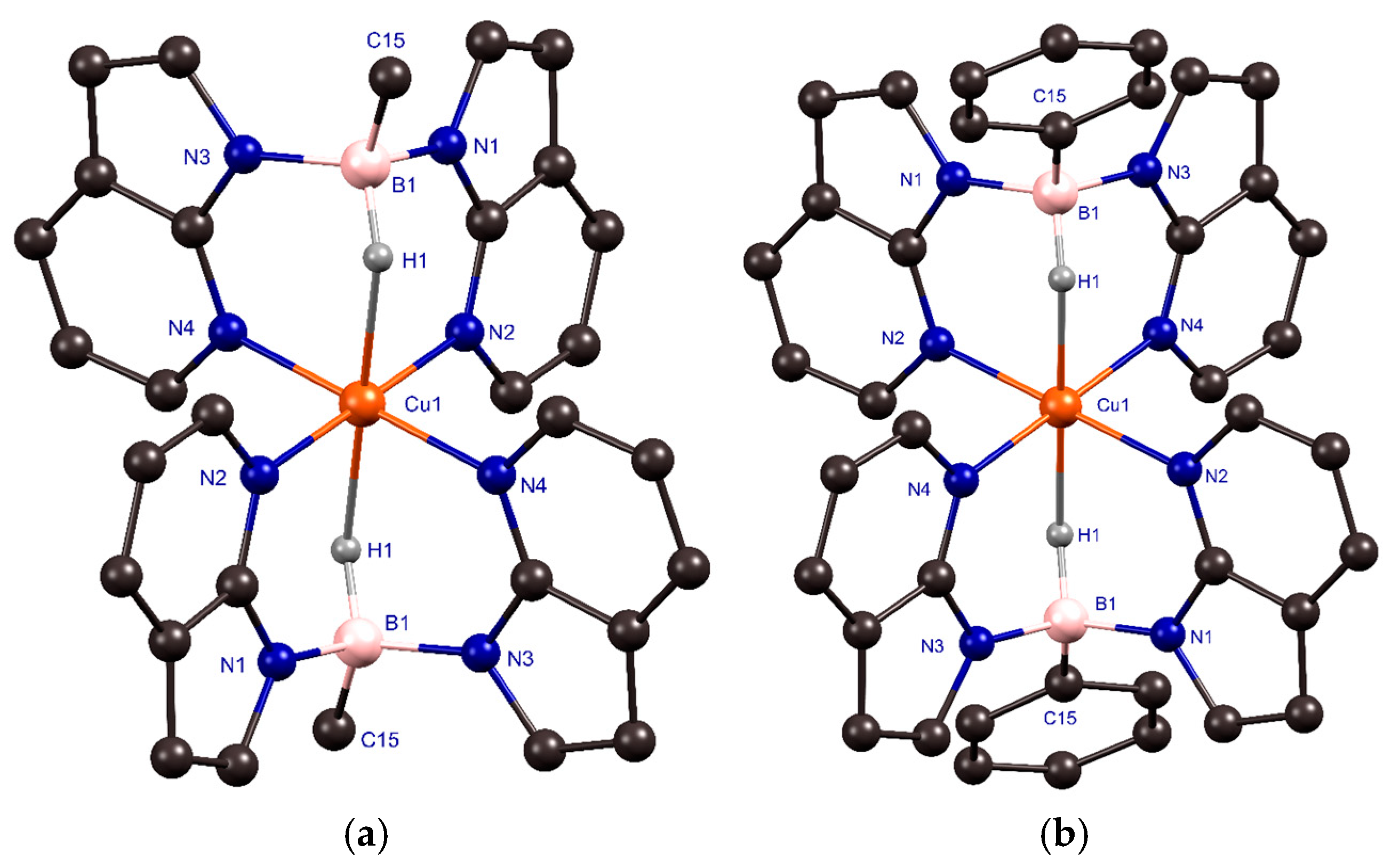
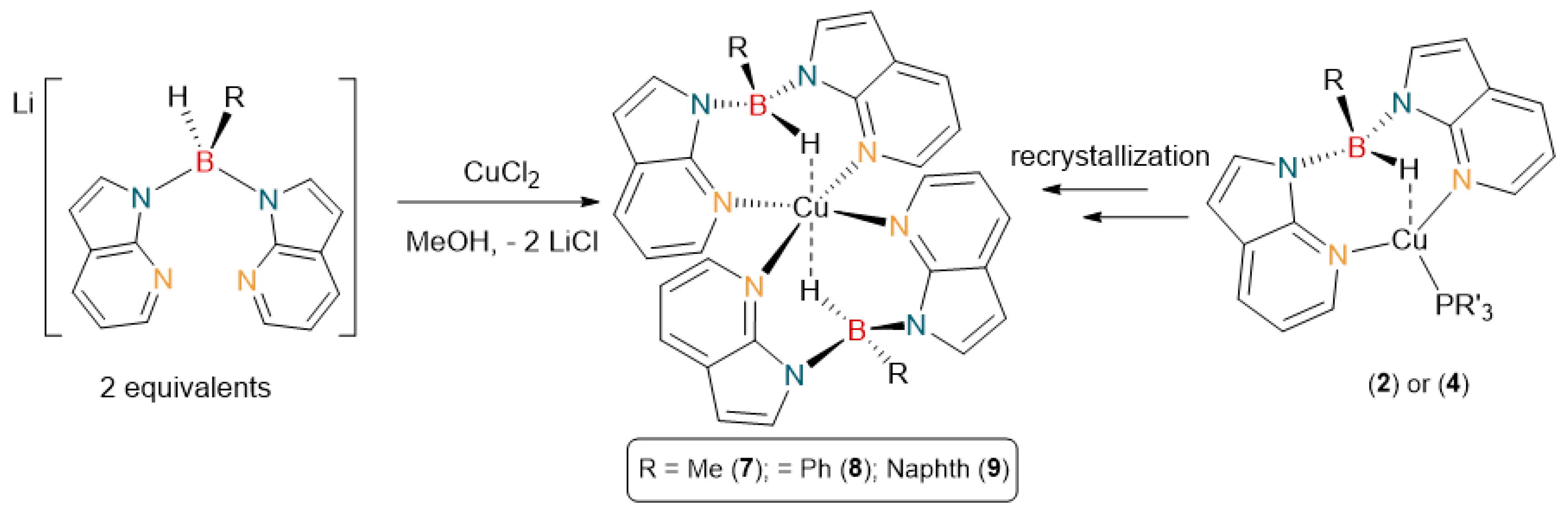
2.3. Synthesis and Crystallization of Copper(II) Complexes
3. Materials and Experimental Methods
3.1. Synthesis of [Cu{κ3-N,N,H-MeBai}(PPh3)] (1)
3.2. Synthesis of [Cu{κ3-N,N,H-PhBai}(PPh3)] (2)
3.3. Synthesis of Synthesis of [Cu{κ3-N,N,H-NaphthBai}(PPh3)] (3)
3.4. Synthesis of [Cu{κ3-N,N,H-MeBai}(PCy3)] (4)
3.5. Synthesis of [Cu{κ3-N,N,H-PhBai}(PCy3)] (5)
3.6. Synthesis of [Cu{κ3-N,N,H-NaphthBai}(PCy3)] (6)
3.7. Synthesis of [Cu{κ3-N,N,H-MeBai}2] (7)
3.8. Synthesis of [Cu{κ3-N,N,H-PhBai}2] (8)
3.9. Synthesis of [Cu{κ3-N,N,H-NaphthBai}2] (9)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Trofimenko, S. Boron-Pyrazole Chemistry. J. Am. Chem. Soc. 1966, 88, 1842–1844. [Google Scholar] [CrossRef]
- Trofimenko, S. Scorpionates: The Coordination of Poly(pyrazolyl)borate Ligands; Imperial College Press: London, UK, 1999. [Google Scholar]
- Pettinari, C. Scorpionates II: Chelating Borate Ligands; Imperial College Press: London, UK, 2008. [Google Scholar] [CrossRef]
- Pettinari, C. Scorpionate Compounds. Eur. J. Inorg. Chem. 2016, 2016, 2209–2211. [Google Scholar] [CrossRef]
- Muñoz-Molina, J.M.; Belderrain, T.R.; Pérez, P.J. Trispyrazolylborate coinage metals complexes: Structural features and catalytic transformations. Coord. Chem. Rev. 2019, 390, 171–189. [Google Scholar] [CrossRef]
- Tsoureas, N.; Owen, G.R.; Hamilton, A.; Orpen, A.G. Flexible scorpionates for transfer hydrogenation: The first example of their catalytic application. Dalton Trans. 2008, 6039–6044. [Google Scholar] [CrossRef]
- Naktode, K.; Reddy, T.D.N.; Nayek, H.P.; Mallik, B.S.; Panda, T.K. Heavier group 2 metal complexes with a flexible scorpionate ligand based on 2-mercaptopyridine. RSC Adv. 2015, 5, 51413–51420. [Google Scholar] [CrossRef] [Green Version]
- Owen, G.R. Hydrogen atom storage upon Z-class borane ligand functions: An alternative approach to ligand cooperation. Chem. Soc. Rev. 2012, 41, 3535–3546. [Google Scholar] [CrossRef]
- Spicer, M.D.; Reglinski, J. Soft Scorpionate Ligands Based on Imidazole-2-thione Donors. Eur. J. Inorg. Chem. 2009, 2009, 1553–1574. [Google Scholar] [CrossRef]
- Neshat, A.; Shahsavari, H.R.; Mastrorilli, P.; Todisco, S.; Haghighi, M.G.; Notash, B. A Borane Platinum Complex Undergoing Reversible Hydride Migration in Solution. Inorg. Chem. 2018, 57, 1398–1407. [Google Scholar] [CrossRef]
- Nuss, G.; Ozwirk, A.; Harum, B.N.; Saischek, G.; Belaj, F.; Mösch-Zanetti, N.C. Copper Complexes with a Hybrid Scorpionate Ligand Containing Pyridazine-3-thione. Eur. J. Inorg. Chem. 2012, 2012, 4701–4707. [Google Scholar] [CrossRef]
- Kimblin, C.; Bridgewater, B.M.; Hascall, T.; Parkin, G. The synthesis and structural characterization of bis(mercaptoimidazolyl)hydroborato complexes of lithium, thallium and zinc. J. Chem. Soc. Dalton Trans. 2000, 891–897. [Google Scholar] [CrossRef]
- Garner, M.; Reglinski, J.; Cassidy, I.; Spicer, M.D.; Kennedy, A.R. Hydrotris(methimazolyl)borate, a soft analogue of hydrotris(pyrazolyl)borate. Preparation and crystal structure of a novel zinc complex. Chem. Commun. 1996, 1975–1976. [Google Scholar] [CrossRef]
- Dodds, C.A.; Garner, M.; Reglinski, J.; Spicer, M.D. Coinage Metal Complexes of a Boron-Substituted Soft Scorpionate Ligand. Inorg. Chem. 2006, 45, 2733–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuss, G.; Saischek, G.; Harum, B.N.; Volpe, M.; Belaj, F.; Mösch-Zanetti, N.C. Pyridazine Based Scorpionate Ligand in a Copper Boratrane Compound. Inorg. Chem. 2011, 50, 12632–12640. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.L.; Tso, K.C.-H.; Cao, B.; Yang, C.; Chen, D.; Chang, X.-Y.; Huang, J.-S.; Che, C.-M. Tripodal S-Ligand Complexes of Copper(I) as Catalysts for Alkene Aziridination, Sulfide Sulfimidation, and C–H Amination. Inorg. Chem. 2017, 56, 4253–4257. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Jia, W.L.; Wu, G.; Wang, S. Cu(i) and Zn(ii) complexes of 7-azaindole-containing scorpionates: Structures, luminescence and fluxionality. Dalton Trans. 2005, 433–438. [Google Scholar] [CrossRef]
- Saha, K.; Ramalakshmi, R.; Gomosta, S.; Pathak, K.; Dorcet, V.; Roisnel, T.; Halet, J.-F.; Ghosh, S. Design, Synthesis, and Chemistry of Bis(σ)borate and Agostic Complexes of Group 7 Metals. Chem. A Eur. J. 2017, 23, 9812–9820. [Google Scholar] [CrossRef]
- Groshens, T.J. Synthesis, characterization, and coordination chemistry of the dihydrobis(5-aminotetrazol-1-yl)borate anion. J. Coord. Chem. 2010, 63, 1882–1892. [Google Scholar] [CrossRef]
- Bailey, P.J.; Bell, N.L.; Gim, L.L.; Yucheng, T.; Funnell, N.; White, F.; Parsons, S. “Twisted” scorpionates: Synthesis of a tris(2-pyridonyl)borate (Thp) ligand; lessons in the requirements for successful B(L2D)3 type ligands. Chem. Commun. 2011, 47, 11659–11661. [Google Scholar] [CrossRef] [Green Version]
- Schinabeck, A.; Rau, N.; Klein, M.; Sundermeyer, J.; Yersin, H. Deep blue emitting Cu(i) tripod complexes. Design of high quantum yield materials showing TADF-assisted phosphorescence. Dalton Trans. 2018, 47, 17067–17076. [Google Scholar] [CrossRef]
- Grätz, M.; Bäcker, A.; Vondung, L.; Maser, L.; Reincke, A.; Langer, R. Donor ligands based on tricoordinate boron formed by B–H-activation of bis(phosphine)boronium salts. Chem. Commun. 2017, 53, 7230–7233. [Google Scholar] [CrossRef]
- MacMillan, S.N.; Harman, W.H.; Peters, J.C. Facile Si–H bond activation and hydrosilylation catalysis mediated by a nickel–borane complex. Chem. Sci. 2014, 5, 590–597. [Google Scholar] [CrossRef] [Green Version]
- Al-Harbi, A.; Rong, Y.; Parkin, G. Synthesis and Structural Characterization of Bis(2-oxoimidazolyl)hydroborato Complexes: A New Class of Bidentate Oxygen-Donor Ligands. Inorg. Chem. 2013, 52, 10226–10228. [Google Scholar] [CrossRef] [PubMed]
- Al-Harbi, A.; Kriegel, B.; Gulati, S.; Hammond, M.J.; Parkin, G. Bis- and Tris(2-oxobenzimidazolyl)hydroborato Complexes of Sodium and Thallium: New Classes of Bidentate and Tridentate Oxygen Donor Ligands. Inorg. Chem. 2017, 56, 15271–15284. [Google Scholar] [CrossRef] [PubMed]
- Landry, V.K.; Buccella, D.; Pang, K.; Parkin, G. Bis- and tris(2-seleno-1-methylimidazolyl)hydroborato complexes, {[BseMe]ZnX}2(X = Cl, I), [BseMe]2Zn and [TseMe]Re(CO)3: Structural evidence that the [BseMe] ligand is not merely a “heavier” version of the sulfur counterpart, [BmMe]. Dalton Trans. 2007, 866–870. [Google Scholar] [CrossRef]
- Holler, S.; Tüchler, M.; Roschger, M.C.; Belaj, F.; Veiros, L.F.; Kirchner, K.; Mösch-Zanetti, N.C. Three-Fold-Symmetric Selenium-Donor Metallaboratranes of Cobalt and Nickel. Inorg. Chem. 2017, 56, 12670–12673. [Google Scholar] [CrossRef]
- Tsoureas, N.; Nunn, J.; Bevis, T.; Haddow, M.F.; Hamilton, A.; Owen, G.R. Strong agostic-type interactions in ruthenium benzylidene complexes containing 7-azaindole based scorpionate ligands. Dalton Trans. 2011, 40, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Tsoureas, N.; Bevis, T.; Butts, C.P.; Hamilton, A.; Owen, G.R. Further Exploring the “Sting of the Scorpion”: Hydride Migration and Subsequent Rearrangement of Norbornadiene to Nortricyclyl on Rhodium(I). Organometallics 2009, 28, 5222–5232. [Google Scholar] [CrossRef]
- Tsoureas, N.; Hope, R.F.; Haddow, M.F.; Owen, G.R. Important Steric Effects Resulting from the Additional Substituent at Boron within Scorpionate Complexes Containing κ3-NNH Coordination Modes. Eur. J. Inorg. Chem. 2011, 2011, 5233–5241. [Google Scholar] [CrossRef]
- Tsoureas, N.; Kuo, Y.-Y.; Haddow, M.F.; Owen, G.R. Double addition of H2 to transition metal–borane complexes: A ‘hydride shuttle’ process between boron and transition metal centres. Chem. Commun. 2011, 47, 484–486. [Google Scholar] [CrossRef] [Green Version]
- Da Costa, R.C.; Rawe, B.W.; Iannetelli, A.; Tizzard, G.J.; Coles, S.J.; Guwy, A.J.; Owen, G.R. Stopping Hydrogen Migration in Its Tracks: The First Successful Synthesis of Group Ten Scorpionate Complexes Based on Azaindole Scaffolds. Inorg. Chem. 2019, 58, 359–367. [Google Scholar] [CrossRef]
- Saito, T.; Kuwata, S.; Ikariya, T. Synthesis and Reactivity of Tris(7-azaindolyl)boratoruthenium Complex. Comparison with Poly(methimazolyl)borate Analogue. Chem. Lett. 2006, 35, 1224–1225. [Google Scholar] [CrossRef]
- Hill, A.F.; Owen, G.R.; White, A.J.P.; Williams, D.J. The sting of the scorpion: A metallaboratrane. Angew. Chem. Int. Ed. 1999, 38, 2759–2761. [Google Scholar] [CrossRef]
- Tsoureas, N.; Haddow, M.F.; Hamilton, A.; Owen, G.R. A new family of metallaboratrane complexes based on 7-azaindole: B–H activation mediated by carbon monoxide. Chem. Commun. 2009, 2538–2540. [Google Scholar] [CrossRef]
- Tsoureas, N.; Hamilton, A.; Haddow, M.F.; Harvey, J.N.; Orpen, A.G.; Owen, G.R. Insight into the Hydrogen Migration Processes Involved in the Formation of Metal–Borane Complexes: Importance of the Third Arm of the Scorpionate Ligand. Organometallics 2013, 32, 2840–2856. [Google Scholar] [CrossRef]
- Da Costa, R.C.; Rawe, B.W.; Tsoureas, N.; Haddow, M.F.; Sparkes, H.A.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Preparation and reactivity of rhodium and iridium complexes containing a methylborohydride based unit supported by two 7-azaindolyl heterocycles. Dalton Trans. 2018, 47, 11047–11057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, G.R.; Tsoureas, N.; Hope, R.F.; Kuo, Y.-Y.; Haddow, M.F. Synthesis and characterisation of group nine transition metal complexes containing new mesityl and naphthyl based azaindole scorpionate ligands. Dalton Trans. 2011, 40, 5906–5915. [Google Scholar] [CrossRef]
- Lobbia, G.G.; Pettinari, C.; Santini, C.; Somers, N.; Skelton, B.; White, A.H. Synthesis, reactivity and solid-state structural studies of new phosphino copper(I) derivatives of hydrotris(3-methyl-2-thioxo-1-imidazolyl)borate. Inorg. Chim. Acta 2001, 319, 15–22. [Google Scholar] [CrossRef]
- Cordero, B.; Gómez, V.; Platero-Prats, A.E.; Revés, M.; Echeverría, J.; Cremades, E.; Barragán, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand/Complex | 31P{1H} NMR | 11B{1H} NMR | h.h.w in 11B/11B{1H} | 1H{11B} NMR B-H | IR (B-H) Powder Film |
|---|---|---|---|---|---|
| Li[MeBai] | - | −8.14 | - | 4.66 | 2396, 2272 |
| Li[PhBai] | - | −6.80 | - | 5.47 | 2394, 2264 |
| Li[NaphthBai] | - | −6.87 | - | 6.05 | 2429, 2272 |
| [Cu(MeBai)(PPh3)] (1) | 1.41 | −7.27 | 186/129 | 5.63 | 2143, 2089 |
| [Cu(PhBai)(PPh3)] (2) | 1.17 | −6.24 | 184/121 | 6.58 | 2120, 2062 |
| [Cu(NaphthBai)(PPh3)] (3) | 1.64 | −6.23 | 180/133 | 6.97 | 2125, 2082 |
| [Cu(MeBai)(PCy3)] (4) | 17.74 | −7.65 | 181/123 | 5.49 | 2141, 2087 |
| [Cu(PhBai)(PCy3)] (5) | 18.08 | −6.37 | 175/129 | 6.36 | 2169, 2076 |
| [Cu(NaphthBai)(PCy3)] (6) | 18.63 | −6.96 | 213/123 | 6.72 | 2189, 2090 |
| Distance (Å)/Angle (°) | 1 | 2 | 3 • (MeCN) | Cu(Tai)(PPh3) a |
|---|---|---|---|---|
| Cu(1)–N(2)/Cu(1)–N(4) | 2.0228(5)/2.0174(5) | 2.0259(5)/2.0206(6) | 2.0350(8)/2.0477(9) | 2.008(3)/2.009(3) |
| B(1)–N(1)/B(1)–N(3) | 1.5507(9)/1.5652(9) | 1.5522(9)/1.5598(10) | 1.5561(14)/1.5567(14) | 1.558(5)/1.547(4)/1.550(4) b |
| B(1)–C(15) | 1.6106(9) | 1.6127(11) | 1.6181(16) | - |
| Cu(1)–P(1) | 2.18341(17) | 2.1825(2) | 2.1899(3) | 2.1844(12) |
| Cu(1)∙∙∙B(1) | 2.7757(7) | 2.7741(8) | 2.7860(14) | 2.808(3) |
| B(1)–H(1) | 1.230(8) | 1.268(9) | 1.282(12) | 1.20(3) |
| Cu(1)∙∙∙H(1) | 1.847(8) | 1.813(9) | 1.803(11) | 1.81(3) |
| N(2)–Cu(1)–P(1) | 125.192(16) | 118.485(15) | 118.79(3) | 125.31(8) |
| N(4)–Cu(1)–P(1) | 123.969(17) | 121.888(19) | 124.43(3) | 126.73(9) |
| N(4)–Cu(1)–N(2) | 107.73(2) | 108.70(2) | 107.27(3) | 105.87(12) |
| H(1)–Cu(1)–P(1) | 104.7(2) | 119.5(3) | 118.3(3) | 107(1) |
| H(1)–Cu(1)–N(2) | 90.7(2) | 88.9(2) | 88.1(3) | 88(1) |
| H(1)–Cu(1)–N(4) | 90.6(2) | 92.0(3) | 91.1(3) | 86(1) |
| ∑ of angles of non-hydrogen substituents at copper | 356.89(3) | 349.07(3) | 350.49(5) | 357.91(17) |
| N(1)–B(1)–N(3) | 109.77(5) | 109.42(6) | 106.89(8) | 111.7(3) |
| C(15)–B(1)–N(1) | 110.77(5) | 114.67(6) | 114.94(9) | 113.7(2) b, c |
| C(15)–B(1)–N(3) | 112.35(5) | 110.53(5) | 111.63(9) | 111.8(3) b, c |
| ∑ of angles of non-hydrogen substituents at boron | 332.89(9) | 334.62(9) | 333.46(15) | 337.2(5) |
| B–H∙∙∙Cu(1) | 127.7(5) | 127.6(6) | 128.3(7) | 136(2) |
| Cu(1)–N–N–B(1) d | −0.75(4)/−2.07(4) | −4.72(4)/−1.55(4) | 7.07(6)/12.13(6) | −5.7(2)/−7.8(2) |
| Position of the Ar group at B e | - | 64.48(8) | 57.66(14) | 178.6(2) b, c |
| Distance (Å)/Angle (°) | 4 | 5 a | 6 • (MeCN) |
|---|---|---|---|
| Cu(1)–N(2)/Cu(1)–N(4) | 2.0740(16)/1.9955(16) | 2.034(3)/2.032(3) | 2.0384(4)/2.0515(4) |
| B(1)–N(1)/B(1)–N(3) | 1.560(3)/1.565(3) | 1.555(5)/1.552(5) | 1.5646(7)/1.5545(7) |
| B(1)–C(15) | 1.603(3) | 1.620(6) | 1.6161(7) |
| Cu(1)–P(1) | 2.1802(5) | [2.205(3)]/[2.208(3)] a | 2.19484(15) |
| Cu(1)∙∙∙B(1) | 2.763(2) | 2.776(4) | 2.7995(7) |
| B(1)–H(1) | 1.23(2) | 1.05(6) | 1.243(7) |
| Cu(1)∙∙∙H(1)B(1) | 1.85(2) | 1.92(6) | 1.847(6) |
| N(2)–Cu(1)–P(1) | 118.87(5) | [118.17(11)]/[129.61(11)] a | 126.875(13) |
| N(4)–Cu(1)–P(1) | 137.38(5) | [129.65(11)]/[118.19(11)] a | 123.195(13) |
| N(4)–Cu(1)–N(2) | 99.77(6) | 109.95(12) | 104.488(17) |
| H(1)–Cu(1)–P(1) | 105.1(7) | [109.1(18)]/[109.1(18)] a | 112.0(2) |
| H(1)–Cu(1)–N(2) | 93.4(6) | 86.9(17) | 88.0(2) |
| H(1)–Cu(1)–N(4) | 88.7(7) | 86.7(17) | 90.1(2) |
| ∑ of angles of non-hydrogen substituents at copper | 356.02(9) | [357.7(2)]/[357.7(2)] a | 354.56(3) |
| N(1)–B(1)–N(3) | 110.42(16) | 110.6(3) | 107.81(4) |
| C(15)–B(1)–N(1) | 111.14(15) | 112.0(3) | 113.59(4) |
| C(15)–B(1)–N(3) | 111.19(17) | 112.1(3) | 112.34(4) |
| ∑ of angles of non-hydrogen substituents at boron | 332.8(3) | 334.7(5) | 333.74(7) |
| B–H∙∙∙Cu(1) | 127.0(16) | 136(5) | 128.8(5) |
| Cu(1)–N–N–B(1) b | 12.24(11)/20.97(11) | 1.7(2)/−1.7(2) | −7.41(3)/−8.30(3) |
| Position of the Ar group at B c | - | 87.5(4) | 70.00(6) |
| Distance (Å)/Angle (°) | 7 | 8 • (CH2Cl2) a |
|---|---|---|
| Cu(1)–N(2)/Cu(1)–N(4) | 2.0161(19)/2.0316(16) | [2.0455(13)/2.0235(12)]/[2.0358(12)/2.0159(12)] |
| B(1)–N(1)/B(1)–N(3) | 1.560(3)/1.553(3) | [1.547(2)/1.554(2)]/[1.5478(19)/1.554(2)] |
| B(1)–C(15) | 1.606(3) | [1.599(2)]/[1.603(2)] |
| Cu(1)∙∙∙B(1) | 2.944(2) | [2.9384(16)]/[2.9281(15)] |
| B(1)–H(1) | 1.18(2) | [1.16(2)]/[1.14(2)] |
| Cu(1)∙∙∙H(1)B(1) | 2.08(2) | [2.00(2)]/[2.03(2)] |
| N(2)–Cu(1)–N(4) | 89.77(6) | [92.94(5)]/[91.10(5)] |
| N(2)`–Cu(1)–N(4) | 90.23(6) | [87.06(5)]/[88.90(5)]] |
| H(1)–Cu(1)–N(2) | 87.6(6) | [85.0(6)]/[85.1(6)] |
| H(1)–Cu(1)–N(4) | 88.1(6) | [85.3(6)]/[86.3(6)] |
| N(1)–B(1)–N(3) | 107.99(16) | [108.88(12)]/[108.58(11)] |
| C(15)–B(1)–N(1) | 111.40(17) | [112.15(12)]/[114.06(11)] |
| C(15)–B(1)–N(3) | 111.98(16) | [115.15(12)]/[112.51(11)] |
| ∑ of angles of non-hydrogen substituents at boron | 331.4(3) | [336.2(2)]/[335.15(19)] |
| B–H∙∙∙Cu(1) | 126.5(16) | [135.2(15)]/[133.7(15)] |
| Cu(1)–N–N–B(1) b | 0.31(11)/−16.58(12) | [5.73(9)/−7.65(9)]/[−9.25(8)/6.28(8)] |
| Position of the Ar group at B c | - | [77.96(15)]/[78.03(15)] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, M.; Thomas, S.D.; Tizzard, G.J.; Coles, S.J.; Owen, G.R. Synthesis and Structural Characterization of Copper Complexes Containing “R-Substituted” Bis-7-Azaindolyl Borate Ligands. Molecules 2023, 28, 4825. https://doi.org/10.3390/molecules28124825
Jackson M, Thomas SD, Tizzard GJ, Coles SJ, Owen GR. Synthesis and Structural Characterization of Copper Complexes Containing “R-Substituted” Bis-7-Azaindolyl Borate Ligands. Molecules. 2023; 28(12):4825. https://doi.org/10.3390/molecules28124825
Chicago/Turabian StyleJackson, Miriam, Simon D. Thomas, Graham J. Tizzard, Simon J. Coles, and Gareth R. Owen. 2023. "Synthesis and Structural Characterization of Copper Complexes Containing “R-Substituted” Bis-7-Azaindolyl Borate Ligands" Molecules 28, no. 12: 4825. https://doi.org/10.3390/molecules28124825