

Synthesis and In Vitro Biological Evaluation of p-Carborane-Based Di-tert-butylphenol Analogs
 , , , , and
, , , , and
Abstract
:
1. Introduction
2. Results and Discussion
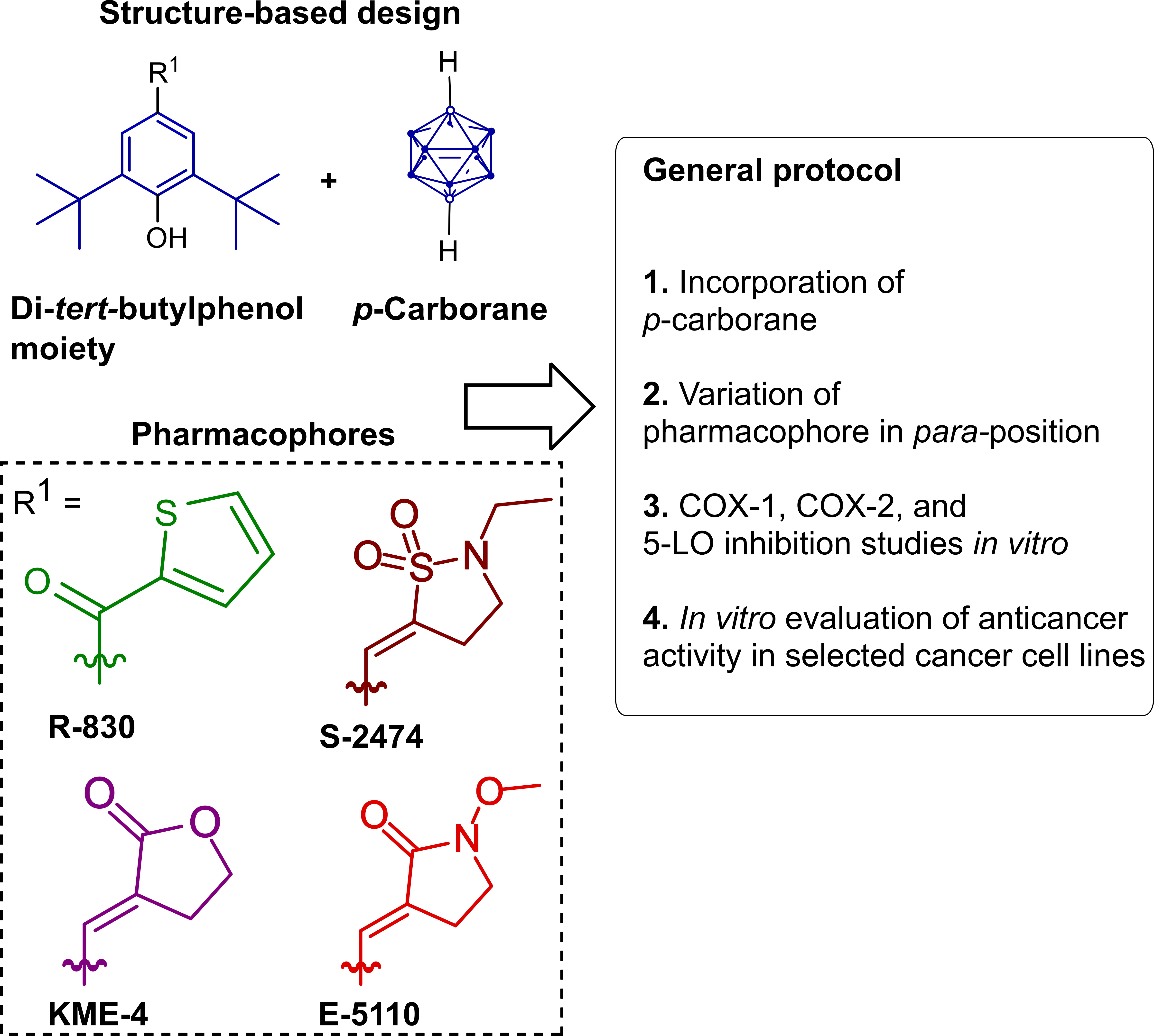
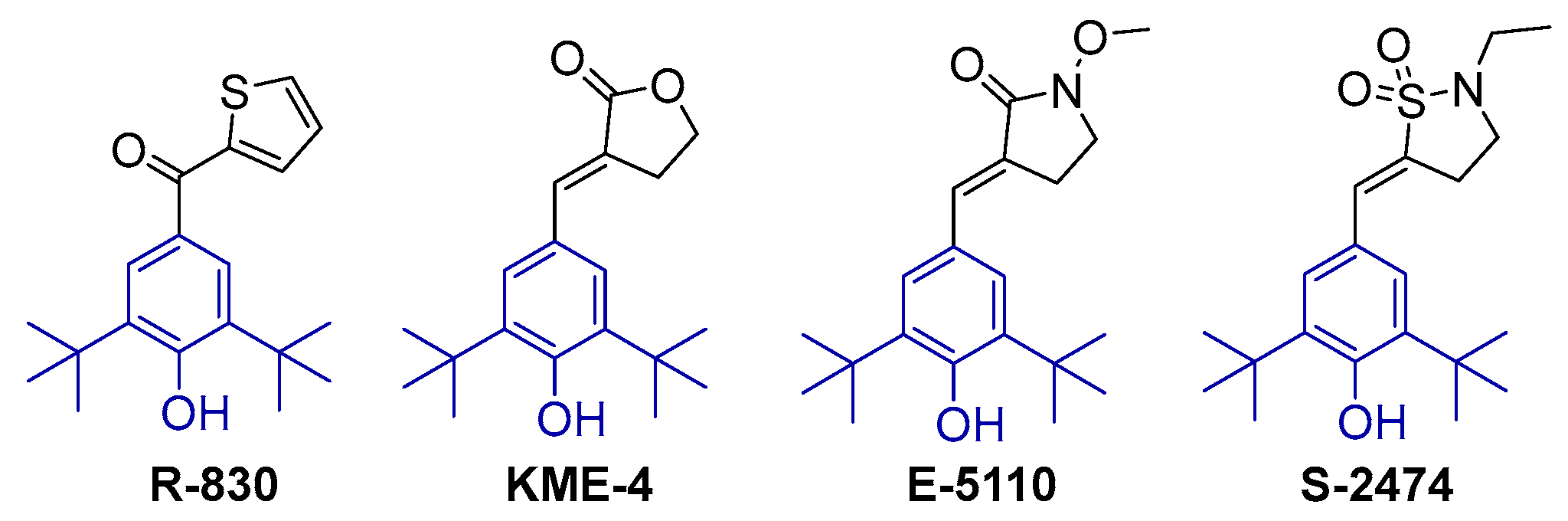
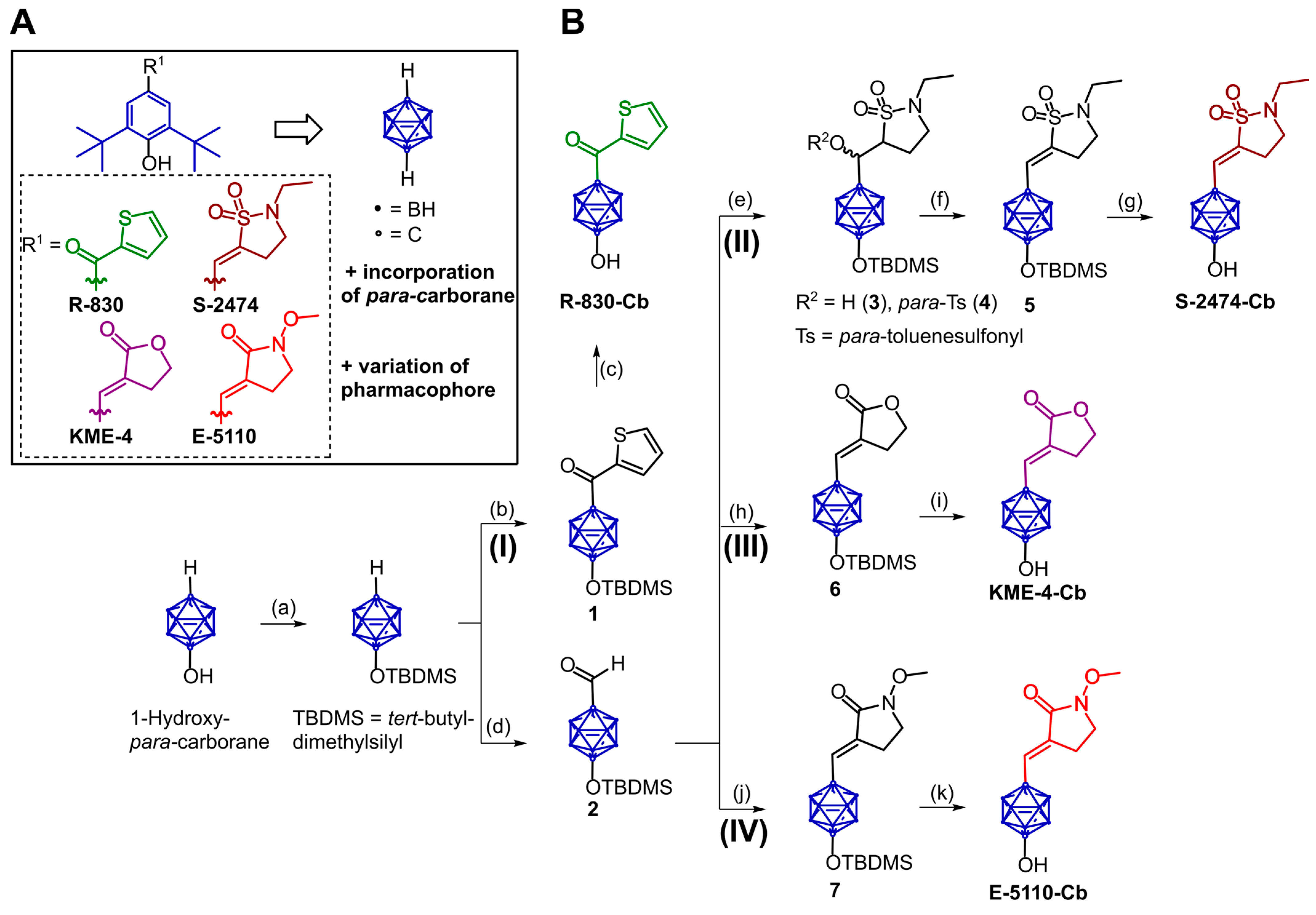
2.1. Design and Synthesis of Di-tert-butylphenol Analogs
2.2. Determination of Lipophilicity (logD) by HPLC
2.3. Evaluation of Inhibitory Potential toward COX
2.4. Evaluation of Inhibitory Potential toward 5-LO
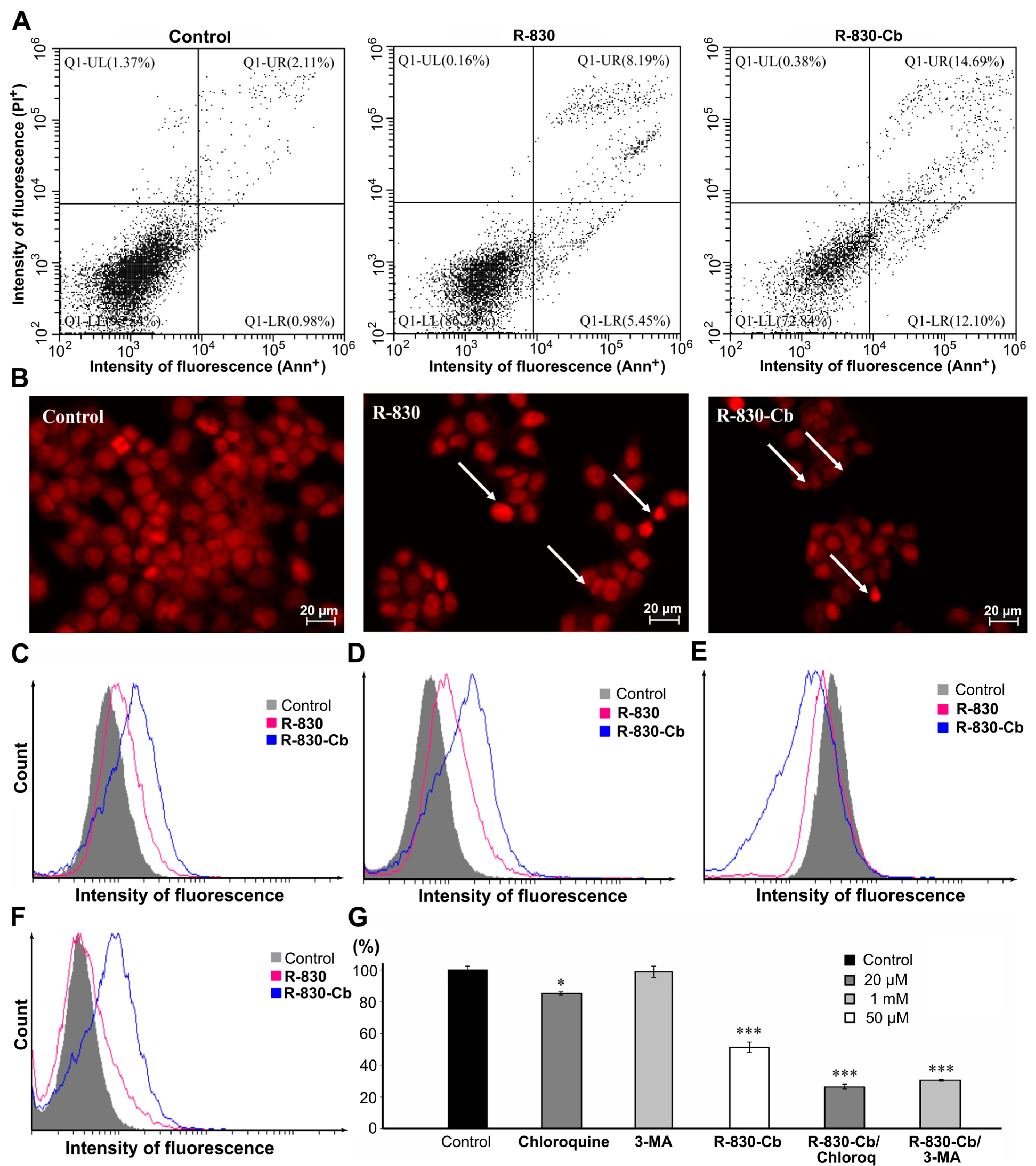
2.5. In Vitro Determination of Cell Viability
3. Experimental Section
3.1. Materials and Methods
3.2. Synthetic Procedures
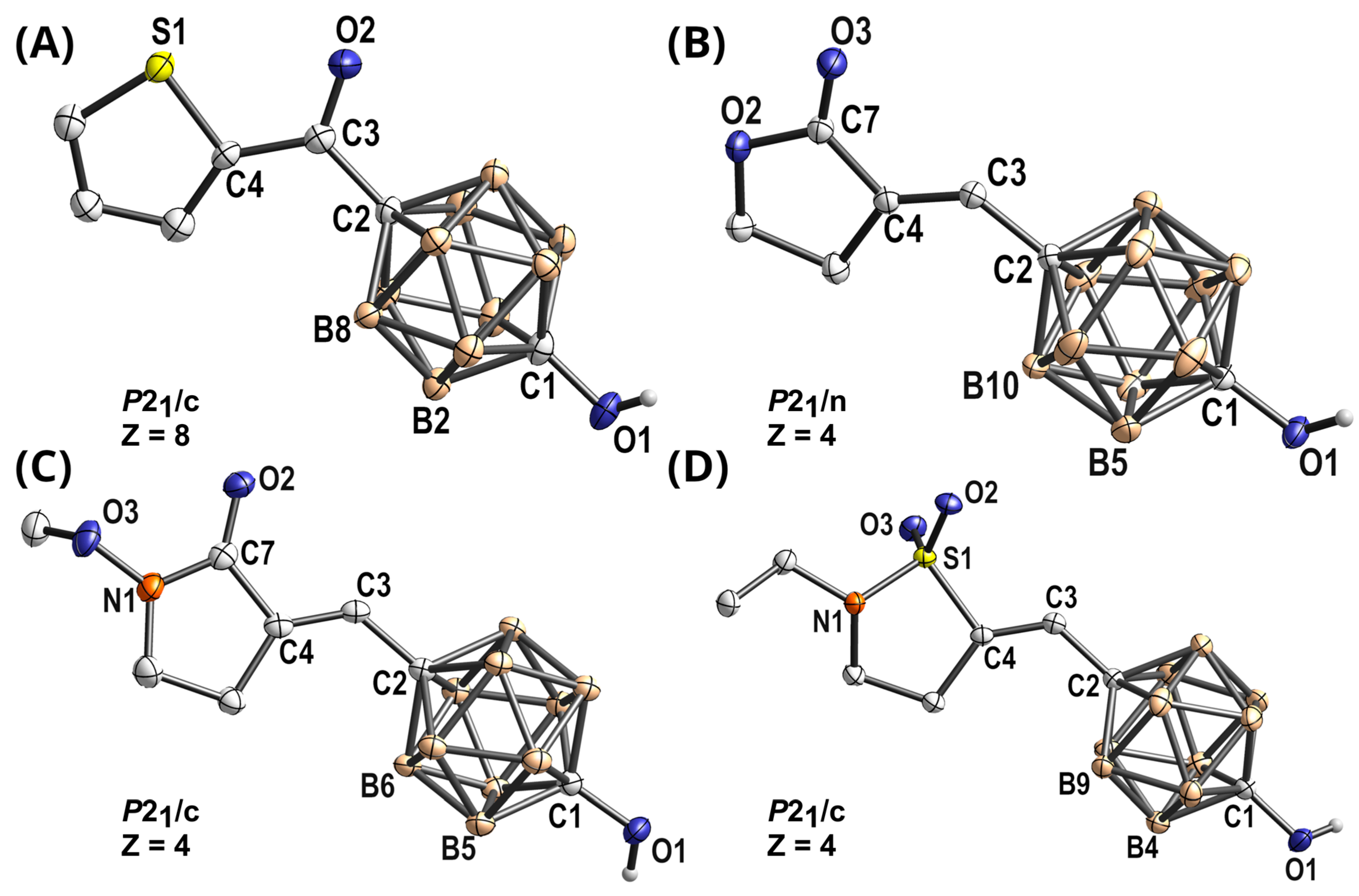
3.2.1. 1-(tert-Butyl-dimethylsiloxy)-12-(thiophen-2′-carbonyl)-1,12-dicarba-closo-dodecaborane(12) (1)
3.2.2. 1-Hydroxy-12-(thiophen-2′-carbonyl)-1,12-dicarba-closo-dodecaborane(12) (R-830-Cb)
3.2.3. 1-(tert-Butyl-dimethylsiloxy)-12-formyl-1,12-dicarba-closo-dodecaborane(12) (2)
3.2.4. 1-(tert-Butyl-dimethylsiloxy)-12-(hydroxymethyl-[2′-ethylisothiazolidine-1′,1′-dioxide])-1,12-dicarba-closo-dodecaborane(12) (3)
3.2.5. 1-(tert-Butyl-dimethylsiloxy)-12-(p-toluenesulfonylmethyl-[2′-ethylisothiazolidine-1′,1′-dioxide])-1,12-dicarba-closo-dodecaborane(12) (4)
3.2.6. (3E)-1-(tert-Butyl-dimethylsiloxy)-12-(methylene-[2′-ethylisothiazolidine-1′,1′-dioxide])-1,12-dicarba-closo-dodecaborane(12) (5)
3.2.7. (3E)-1-Hydroxy-12-(methylene-[2′-ethylisothiazolidine-1′,1′-dioxide])-1,12-dicarba-closo-dodecaborane(12) (S-2474-Cb)
3.2.8. (3E)-1-(tert-Butyldimethylsiloxy)-12-(methylene-[dihydrofurane-2′(3H)-one])-1,12-dicarba-closo-dodecaborane(12) (6)
3.2.9. (3E)-1-Hydroxy-12-(methylene-[dihydrofurane-2′(3H)-one])-1,12-dicarba-closo-dodecaborane(12) (KME-4-Cb)
3.2.10. (3E)-1-(tert-Butyl-dimethylsiloxy)-12-(methylene-[1′-methoxypyrrolidine-2′-one])-1,12-dicarba-closo-dodecaborane(12) (7)
3.2.11. (3E)-1-Hydroxy-12-(methylene-[1′-methoxypyrrolidine-2′-one])-1,12-dicarba-closo-dodecaborane(12) (E-5110-Cb)
3.3. HPLC Measurements
3.3.1. Analysis of Purity Ultra Performance Liquid Chromatography (UPLC)
3.3.2. Determination of Lipophilicity (logD) by HPLC
3.4. COX Inhibition Studies
3.5. 5-LO Inhibitory Studies
3.6. Cell Viability Studies—MTT and CV Assays
3.7. Statistical Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| A375 | Human melanoma cell line |
| A549 | Human lung carcinoma cell line |
| AA | Arachidonic acid |
| AnnV | Annexin V |
| AO | Acridine orange |
| Apostat | FITC-conjugated pan-caspase inhibitor |
| ATR-IR | Attenuated total reflection infrared spectroscopy |
| b/br | Broad |
| BWA4C | N-[(E)-3-(3-Phenoxyphenyl)prop-2-enyl]acetohydroxamic acid |
| CDCl3 | Deuterated chloroform |
| CFSE | Carboxyfluorescein N-succinimidyl ester |
| CIs | Confidence intervals |
| COX | Cyclooxygenase |
| COXIBs | COX-2 specific inhibitors |
| CV | Crystal violet |
| CYP | Cytochrome P450 |
| d | Doublet |
| dd | Doublet of doublets |
| ddd | Doublet of doublets of doublets |
| dtd | Doublet of triplets of doublets |
| DHR 123 | Dihydrorhodamine 123 |
| DMAP | 4-Dimethylaminopyridine |
| FLAP | 5-Lipoxygenase-activating protein |
| FT-ICR | Fourier-transform ion cyclotron resonance mass spectrometry |
| GI | Gastrointestinal |
| HCT116 | Human colorectal carcinoma cell line |
| 5-H(p)ETE | 5-Hydroperoxyeicosatetraenoic acid |
| HPLC | High-performance liquid chromatography |
| HR-ESI-MS | High-resolution electrospray ionization mass spectrometry |
| HT29 | Human colorectal adenocarcinoma cell line |
| IC50 | Half-maximal inhibitory concentration |
| logD | Partition coefficient |
| LO | Lipoxygenase |
| LTs | Leukotrienes |
| LTB4 | Leukotriene B4 |
| m | Medium (IR), multiplet (NMR), meta |
| M | Molarity |
| 3-MA | 3-Methyladenine |
| MDA-MB-231 | M. D. Anderson-Metastatic breast-231 triple negative adenocarcinoma cell line |
| m.p. | Melting point |
| MTT | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide |
| nBuLi | n-Butyllithium |
| NEt3 | Triethylamine |
| n.i. | no inhibition |
| NMR | Nuclear magnetic resonance |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| p | para |
| PBS | Phosphate-buffered saline |
| PFA | Paraformaldehyde |
| PGs | Prostaglandins |
| PGE2 | Prostaglandin E2 |
| PGI2 | Prostaglandin I2 |
| PI | Propidium iodide |
| PMNL | Polymorphonuclear leukocytes |
| ppm | Parts per million |
| p-Ts | p-Toluenesulfonyl |
| q | Quartet |
| RF | Retention factor |
| RT | Room temperature |
| s | Strong (IR), singlet (NMR) |
| SC-560 | 5-(4-Chlorophenyl)-1-(4-methoxyphenyl)-3-trifluoromethyl pyrazole |
| SD | Standard deviation |
| SI | Selectivity index |
| t | Triplet |
| td | Triplet of doublets |
| TBAF | Tetra-n-butylammonium fluoride |
| TBDMSCl | Tert-butyldimethylsilyl chloride |
| TLC | Thin-layer chromatography |
| TMS | Tetramethylsilane |
| tR | Retention time |
| TXA2 | Thromboxane A2 |
| UPLC | Ultra-performance liquid chromatography |
| w | Weak |
References
- Ting, A.H.; McGarvey, K.M.; Baylin, S.B. The cancer epigenome-components and functional correlates. Genes Dev. 2006, 20, 3215–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Motilva, V.; Martorell-Calatayud, A.; Nagore, E. Non-steroidal Anti-Inflammatory Drugs and Melanoma. Curr. Pharm. Des. 2012, 18, 3966–3978. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Lessons from HEREDITARY colorectal cancer. Cell 1996, 87, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Prendergast, G.C.; Metz, R.; Muller, A. Towards a genetic definition of cancer-associated inflammation: Role of the IDO pathway. Am. J. Clin. Pathol. 2010, 176, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arneth, B. Tumor Microenvironment. Medicina 2019, 56, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, A.M.; Kleczko, E.K.; Nemenoff, R. Eicosanoids in Cancer: New Roles in Immunoregulation. Front. Pharmacol. 2020, 11, 595498. [Google Scholar] [CrossRef]
- Moore, G.Y.; Pidgeon, G. Cross-Talk between Cancer Cells and the Tumour Microenvironment: The Role of the 5-Lipoxygenase Pathway. Int. J. Mol. Sci. 2017, 18, 236. [Google Scholar] [CrossRef] [Green Version]
- Charlier, C.; Michaux, C. Dual inhibition of cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LOX) as a new strategy to provide safer non-steroidal anti-inflammatory drugs. Eur. J. Med. Chem. 2003, 38, 645–659. [Google Scholar] [CrossRef]
- Jacob, P.J.; Manju, S.L.; Ethiraj, K.R.; Elias, G. Safer anti-inflammatory therapy through dual COX-2/5-LOX inhibitors: A structure-based approach. Eur. J. Pharm. Sci. 2018, 121, 356–381. [Google Scholar]
- Brücher, B.L.D.M.; Jamall, I.S. Eicosanoids in carcinogenesis. 4open 2019, 2, 9. [Google Scholar] [CrossRef]
- Sharma, V.; Bhatia, P.; Alam, O.; Naim, M.J.; Nawaz, F.; Sheikh, A.A.; Jha, M. Recent advancement in the discovery and development of COX-2 inhibitors: Insight into biological activities and SAR studies (2008–2019). Bioorg. Chem. 2019, 89, 103007. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.; Ismail, S.M.; Zarghi, A. Selective COX-2 inhibitors as anticancer agents: A patent review (2014–2018). Expert Opin. Ther. Pat. 2019, 29, 407–427. [Google Scholar] [CrossRef]
- Mohsin, N.-A.; Irfan, M. Selective cyclooxygenase-2 inhibitors: A review of recent chemical scaffolds with promising anti-inflammatory and COX-2 inhibitory activities. Med. Chem. Res. 2020, 29, 809–830. [Google Scholar] [CrossRef]
- Roos, J.; Grösch, S.; Werz, O.; Schröder, P.; Ziegler, S.; Fulda, S.; Paulus, P.; Urbschat, A.; Kühn, B.; Maucher, I.; et al. Regulation of tumorigenic Wnt signaling by cyclooxygenase-2, 5-lipoxygenase and their pharmacological inhibitors: A basis for novel drugs targeting cancer cells? Pharmacol. Ther. 2016, 157, 43–64. [Google Scholar] [CrossRef]
- Meshram, M.A.; Bhise, U.O.; Makhal, P.N.; Kaki, V. Synthetically-tailored and nature-derived dual COX-2/5-LOX inhibitors: Structural aspects and SAR. Eur. J. Med. Chem. 2021, 225, 113804. [Google Scholar] [CrossRef]
- Arias-Negrete, S.; Keller, K.; Chadee, K. Proinflammatory cytokines regulate cyclooxygenase-2 mRNA expression in human macrophages. Biochem. Biophys. Res. Commun. 1995, 208, 582–589. [Google Scholar] [CrossRef]
- Vecchio, A.J.; Simmons, D.M.; Malkowski, M. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 2010, 285, 22152–22163. [Google Scholar] [CrossRef] [Green Version]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Sci. 2011, 10, 655. [Google Scholar]
- Goossens, L.; Pommery, N.; Hénichart, J. COX-2/5-LOX dual acting anti-inflammatory drugs in cancer chemotherapy. Curr. Top. Med. Chem. 2007, 7, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Haase-Kohn, C.; Laube, M.; Donat, C.K.; Belter, B.; Pietzsch, J. CRISPR/Cas9 Mediated Knockout of Cyclooxygenase-2 Gene Inhibits Invasiveness in A2058 Melanoma Cells. Cells 2022, 11, 749. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Belvisi, M.G.; Akarasereenont, P.; Robbins, R.A.; Kwon, O.J.; Croxtall, J.; Barnes, P.J.; Vane, J. Induction of cyclo-oxygenase-2 by cytokines in human pulmonary epithelial cells: Regulation by dexamethasone. Br. J. Pharmacol. 1994, 113, 1008–1014. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.; Krishnan, A.V.; Swami, S.; Nonn, L.; Peehl, D.M.; Feldman, D. Regulation of prostaglandin metabolism by calcitriol attenuates growth stimulation in prostate cancer cells. Cancer Res. 2005, 65, 7917–7925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, M.; Bekeschus, S.; Weltmann, K.-D.; Woedtke, T.; Wende, K. Non-steroidal anti-inflammatory drugs: Recent advances in the use of synthetic COX-2 inhibitors. RSC Med. Chem. 2022, 13, 471–496. [Google Scholar] [CrossRef]
- Orlando, B.J.; Lucido, M.J.; Malkowski, M. The structure of ibuprofen bound to cyclooxygenase-2. J. Struct. Biol. 2015, 189, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Lucido, M.J.; Orlando, B.J.; Vecchio, A.J.; Malkowsk, M. Crystal Structure of Aspirin-Acetylated Human Cyclooxygenase-2: Insight into the Formation of Products with Reversed Stereochemistry. Biochemistry 2016, 55, 1226–1238. [Google Scholar] [CrossRef] [Green Version]
- Haeggström, J.Z.; Funk, C. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef]
- Reddy, K.K.; Rajan, V.K.V.; Gupta, A.; Aparoy, P.; Reddanna, P. Exploration of binding site pattern in arachidonic acid metabolizing enzymes, Cyclooxygenases and Lipoxygenases. BMC Res. Notes 2015, 8, 152. [Google Scholar] [CrossRef] [Green Version]
- Reddy, S.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2013, 6, 41–47. [Google Scholar] [CrossRef] [Green Version]
- Pidgeon, G.; Cathcart, M.-C. The Role of Cyclooxygenases and Lipoxygenases in the Regulation of Tumor Angiogensis; Taylor & Francis: New York, NY, USA, 2013; Chapter 7. [Google Scholar]
- Sinha, S.; Doble, M.; Manju, S. 5-Lipoxygenase as a drug target: A review on trends in inhibitors structural design, SAR and mechanism based approach. Bioorg. Med. Chem. 2019, 27, 3745–3759. [Google Scholar] [CrossRef]
- Orafaie, A.; Mousavian, M.; Orafai, H.; Sadeghian, H. An overview of lipoxygenase inhibitors with approach of in vivo studies. Prostaglandins Other Lipid Mediat. 2020, 148, 106411. [Google Scholar] [CrossRef] [PubMed]
- de Leval, X.; Julemont, F.; Delarge, J.; Pirotte, B.; Dogne, J.-M. New trends in dual 5-LOX/COX inhibition. Curr. Med. Chem. 2002, 9, 941–962. [Google Scholar] [CrossRef] [PubMed]
- Julémont, F.; Dogné, J.-M.; Pirotte, B.; de Leval, X. Recent development in the field of dual COX / 5-LOX inhibitors. Mini-Rev. Med. Chem. 2004, 4, 633–638. [Google Scholar] [CrossRef]
- Le Filliatre, G.; Sayah, S.; Latournerie, V.; Renaud, J.F.; Finet, M.; Hanf, R. Cyclooxygenase and lipoxygenase pathways in mast cell dependent-neurogenic inflammation induced by electrical stimulation of the rat saphenous nerve. Br. J. Pharmacol. 2001, 132, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- He, C.; Wu, Y.; Lai, Y.; Cai, Z.; Liu, Y.; Lai, L. Dynamic eicosanoid responses upon different inhibitor and combination treatments on the arachidonic acid metabolic network. Mol. BioSystems 2012, 8, 1585–1594. [Google Scholar] [CrossRef] [PubMed]
- Busse, W.W. Leukotrienes and Inflammation. Am. J. Respir. Crit. Care. Med. 1996, 157, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Mahboubi-Rabbani, M.; Zarghi, A. Lipoxygenase Inhibitors as Cancer Chemopreventives: Discovery, Recent Developments and Future Perspectives. Curr. Med. Chem. 2021, 28, 1143–1175. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.; Brogden, R.N. Nimesulide. Drugs 1994, 48, 431–454. [Google Scholar] [CrossRef]
- Khanapure, S.P.; Garvey, D.S.; Janero, D.R.; Letts, L.G. Eicosanoids in Inflammation: Biosynthesis, Pharmacology, and Therapeutic Frontiers. Curr. Top. Med. Chem. 2007, 7, 311–340. [Google Scholar] [CrossRef]
- Braeckmann, R.A.; Granneman, G.R.; Locke, C.S.; Machinist, J.M.; Cavanaugh, J.H.; Awni, W. The Pharmacokinetics of Zileuton in Healthy Young and Elderly Volunteers. Clin. Pharmacokinet. 1995, 29, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Paredes, Y.; Massicotte, F.; Pelletier, J.-P.; Martel-Pelletier, J.; Laufer, S.; Lajeunesse, D. Study of the role of leukotriene B4 in abnormal function of human subchondral osteoarthritis osteoblasts: Effects of cyclooxygenase and/or 5-lipoxygenase inhibition. Arthritis Rheumatol. 2002, 46, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Martel-Pelletier, J.; Lajeunesse, D.; Reboul, P.; Pelletier, J.-P. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann. Rheum. Dis. 2003, 62, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, D.V.; Fernandes, J.C.; Martel-Pelletier, J.; Jolicoeur, F.-C.; Reboul, P.; Laufer, S.; Tries, S.; Pelletier, J.-P. In vivo dual inhibition of cyclooxygenase and lipoxygenase by ML-3000 reduces the progression of experimental osteoarthritis: Suppression of collagenase 1 and interleukin-1? synthesis. Arthritis Rheum. 2001, 44, 2320–2330. [Google Scholar] [CrossRef]
- Ruiz, J. QSAR study of dual cyclooxygenase and 5-lipoxygenase inhibitors 2,6-di-tert-butylphenol derivatives. Bioorg. Med. Chem. 2003, 11, 4207–4216. [Google Scholar] [CrossRef]
- Moore, G.G.; Swingle, K.F. 6-Di-tert-butyl-4-(2′-thenoyl)phenol(R-830): A novel nonsteroidal anti-inflammatory agent with antioxidant properties. Agents Actions 1982, 12, 674–683. [Google Scholar] [CrossRef]
- Blackham, A.; Norris, A.A.; Woods, F. Models for evaluating the anti-inflammatory effects of inhibitors of arachidonic acid metabolism. J. Pharm. Pharmacol. 1985, 37, 787–793. [Google Scholar] [CrossRef]
- Hidaka, T.; Hosoe, K.; Ariki, Y.; Takeo, K.; Yamashita, T.; Katsumi, I.; Kondo, H.; Yamashita, K.; Watanabe, K. Pharmacological properties of a new anti-inflammatory compound, alpha-(3,5-di-tert-butyl-4-hydroxybenzylidene)-gamma-butyrolacto ne (KME-4), and its inhibitory effects on prostaglandin synthetase and 5-lipoxygenase. Jpn. J. Clin. Pharmacol. Ther. 1984, 36, 77–85. [Google Scholar]
- Hidaka, T.; Takeo, K.; Hosoe, K.; Katsumi, I.; Yamashita, T.; Watanabe, K. Inhibition of polymorphonuclear leukocyte 5-lipoxygenase and platelet cyclooxygenase by alpha-(3,5-di-tert-butyl-4-hydroxybenzylidene)-gamma-butyrolacto ne (KME-4), a new anti-inflammatory drug. Jpn. J. Clin. Pharmacol. Ther. 1985, 38, 267–272. [Google Scholar]
- Hidaka, T.; Hosoe, K.; Katsumi, I.; Yamashita, T.; Watanabe, K. The effect of alpha-(3,5-di-t-butyl-4-hydroxybenzylidene)-gamma-butyrolactone (KME-4), a new anti-inflammatory drug, on leucocyte migration in rat carrageenan pleurisy. J Pharm Pharmacol. 1986, 38, 244–247. [Google Scholar] [CrossRef]
- Hidaka, T.; Hosoe, K.; Yamashita, T.; Watanabe, K. Effect of alpha-(3,5-di-tert-butyl-4-hydroxybenzylidene)-gamma-butyrolactone (KME-4), a new anti-inflammatory drug, on the established adjuvant arthritis in rats. Jpn. J. Clin. Pharmacol. Ther. 1986, 42, 181–187. [Google Scholar]
- Katsumi, I.; Kondo, H.; Yamashita, K.; Hidaka, T.; Hosoe, K.; Yamashita, T.; Watanabe, K. Studies on styrene derivatives. I. Synthesis and antiinflammatory activities of alpha-benzylidene-gamma-butyrolactone derivatives. Chem. Pharm. Bull. 1986, 34, 121–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikuta, H.; Shirota, H.; Kobayashi, S.; Yamagishi, Y.; Yamada, K.; Yamatsu, I.; Katayama, K. Synthesis and antiinflammatory activities of 3-(3,5-di-tert-butyl-4-hydroxybenzylidene)pyrrolidin-2-ones. J. Med. Chem. 1987, 30, 1995–1998. [Google Scholar] [CrossRef] [PubMed]
- Shirota, H.; Goto, M.; Hashida, R.; Yamatsu, I.; Katayama, K. Inhibitory effects of E-5110 on interleukin-1 generation from human monocytes. Agents Actions 1989, 27, 322–324. [Google Scholar] [CrossRef]
- Shirota, H.; Katayama, K.; Ono, H.; Chiba, K.; Kobayashi, S.; Terato, K.; Ikuta, H.; Yamatsu, I. Pharmacological properties of N-methoxy-3-(3,5-ditert-butyl-4-hydroxybenzylidene)-2-pyrrolidone (E-5110), a novel nonsteroidal antiinflammatory agent. Agents Actions 1987, 21, 250–252. [Google Scholar] [CrossRef]
- Katayama, K.; Shirota, H.; Kobayashi, S.; Terato, K.; Ikuta, H.; Yamatsu, I. In vitro effect of N-methoxy-3-(3,5-ditert-butyl-4-hydroxybenzylidene)-2-pyrrolidon e (E-5110), a novel nonsteroidal antiinflammatory agent, on generation of some inflammatory mediators. Agents Actions 1987, 21, 269–271. [Google Scholar] [CrossRef]
- Inagaki, M.; Tsuri, T.; Jyoyama, H.; Ono, T.; Yamada, K.; Kobayashi, M.; Hori, Y.; Arimura, A.; Yasui, K.; Ohno, K.; et al. Novel antiarthritic agents with 1,2-isothiazolidine-1,1-dioxide (gamma-sultam) skeleton: Cytokine suppressive dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase. J. Med. Chem. 2000, 43, 2040–2048. [Google Scholar] [CrossRef]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as pharmacophores: Properties, synthesis, and application strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef]
- Stockmann, P.; Gozzi, M.; Kuhnert, R.; Sárosi, M.B.; Hey-Hawkins, E. New keys for old locks: Carborane-containing drugs as platforms for mechanism-based therapies. Chem. Soc. Rev. 2019, 48, 3497–3512. [Google Scholar] [CrossRef] [Green Version]
- Marfavi, A.; Kavianpour, P.; Rendina, L. Carboranes in drug discovery, chemical biology and molecular imaging. Nat. Rev. Chem. 2022, 6, 486–504. [Google Scholar] [CrossRef]
- Messner, K.; Vuong, B.; Tranmer, G. The Boron Advantage: The Evolution and Diversification of Boron’s Applications in Medicinal Chemistry. Pharmaceuticals 2022, 15, 264. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R. Building Bridges between Inorganic and Organic Chemistry (Nobel Lecture). Angew. Chem. Int. Ed. 1982, 21, 711–724. [Google Scholar] [CrossRef]
- Issa, F.; Kassiou, M.; Rendina, L. Boron in drug discovery: Carboranes as unique pharmacophores in biologically active compounds. Chem. Rev. 2011, 111, 5701–5722. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Du, F.; Tang, L.; Xu, J.; Zhao, Y.; Wu, X.; Li, M.; Shen, J.; Wen, Q.; Cho, C.H.; et al. Carboranes as unique pharmacophores in antitumor medicinal chemistry. Mol. Ther. Oncolytics 2022, 24, 400–416. [Google Scholar] [CrossRef]
- Ali, F.; Hosmane, N.; Zhu, Y. Boron Chemistry for Medical Applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [Green Version]
- Bregadze, V.I. Dicarba-closo-dodecaboranes C2B10H12 and their derivatives. Chem. Rev. 1992, 92, 209–223. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 2nd ed.; Elsevier Science & Technology: Saint Louis, MO, USA, 2011. [Google Scholar]
- Zargham, E.O.; Mason, C.A.; Lee, M. The Use of Carboranes in Cancer Drug Development. Int. J. Cancer Clin. Res. 2019, 6, 110. [Google Scholar]
- Das, B.C.; Nandwana, N.K.; Das, S.; Nandwana, V.; Shareef, M.A.; Das, Y.; Saito, M.; Weiss, L.M.; Almaguel, F.; Hosmane, N.S.; et al. Boron Chemicals in Drug Discovery and Development: Synthesis and Medicinal Perspective. Molecules 2022, 27, 2615. [Google Scholar] [CrossRef]
- Neumann, W.; Xu, S.; Sárosi, M.B.; Scholz, M.S.; Crews, B.C.; Ghebreselasie, K.; Banerjee, S.; Marnett, L.J.; Hey-Hawkins, E. nido-Dicarbaborate Induces Potent and Selective Inhibition of Cyclooxygenase-2. ChemMedChem 2016, 11, 175–178. [Google Scholar] [CrossRef] [Green Version]
- Buzharevski, A.; Paskas, S.; Laube, M.; Lönnecke, P.; Neumann, W.; Murganic, B.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; Hey-Hawkins, E. Carboranyl Analogues of Ketoprofen with Cytostatic Activity against Human Melanoma and Colon Cancer Cell Lines. ACS Omega 2019, 4, 8824–8833. [Google Scholar] [CrossRef] [Green Version]
- Buzharevski, A.; Paskas, S.; Sárosi, M.-B.; Laube, M.; Lönnecke, P.; Neumann, W.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; Hey-Hawkins, E. Carboranyl Analogues of Celecoxib with Potent Cytostatic Activity against Human Melanoma and Colon Cancer Cell Lines. ChemMedChem 2019, 14, 315–321. [Google Scholar] [CrossRef]
- Kuhnert, R.; Sárosi, M.-B.; George, S.; Lönnecke, P.; Hofmann, B.; Steinhilber, D.; Steinmann, S.; Schneider-Stock, R.; Murganić, B.; Mijatović, S.; et al. Carborane-Based Analogues of 5-Lipoxygenase Inhibitors Co-inhibit Heat Shock Protein 90 in HCT116 Cells. ChemMedChem 2019, 14, 255–261. [Google Scholar] [CrossRef]
- Buzharevski, A.; Paskaš, S.; Sárosi, M.-B.; Laube, M.; Lönnecke, P.; Neumann, W.; Murganić, B.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; et al. Carboranyl Derivatives of Rofecoxib with Cytostatic Activity against Human Melanoma and Colon Cancer Cells. Sci. Rep. 2020, 10, 4827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Useini, L.; Mojić, M.; Laube, M.; Lönnecke, P.; Dahme, J.; Sárosi, M.B.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; Hey-Hawkins, E. Carboranyl Analogues of Mefenamic Acid and Their Biological Evaluation. ACS Omega 2022, 7, 24282–24291. [Google Scholar] [CrossRef] [PubMed]
- Useini, L.; Mojić, M.; Laube, M.; Lönnecke, P.; Mijatović, S.; Maksimović-Ivanić, D.; Pietzsch, J.; Hey-Hawkins, E. Carborane Analogues of Fenoprofen Exhibit Improved Antitumor Activity. ChemMedChem 2022, 18, e202200583. [Google Scholar] [CrossRef] [PubMed]
- Braun, S.; Paskaš, S.; Laube, M.; George, S.; Hofmann, B.; Lönnecke, P.; Steinhilber, D.; Pietzsch, J.; Mijatović, S.; Maksimović-Ivanić, D.; et al. In Vitro Cytostatic Effect on Tumor Cells by Carborane-Based Dual Cyclooxygenase-2 and 5-Lipoxygenase Inhibitors. Adv. Ther. 2023, 6, 2200252. [Google Scholar] [CrossRef]
- Braun, S.; Paskaš, S.; Laube, M.; George, S.; Hofmann, B.; Lönnecke, P.; Steinhilber, D.; Pietzsch, J.; Mijatović, S.; Maksimović-Ivanić, D.; et al. Carborane-based Tebufelone Analogs and their Biological Evaluation In Vitro. ChemMedChem 2023, 18, e202300206. [Google Scholar] [CrossRef]
- Ohta, K.; Goto, T.; Yamazaki, H.; Pichierri, F.; Endo, Y. Facile and efficient synthesis of C-hydroxycarboranes and C,C′-dihydroxycarboranes. Inorg. Chem. 2007, 46, 3966–3970. [Google Scholar] [CrossRef]
- Fournier, J.; Arseniyadis, S.; Cossy, J. A modular and scalable one-pot synthesis of polysubstituted furans. Angew. Chem. Int. 2012, 51, 7562–7566. [Google Scholar] [CrossRef]
- Donovan, S.F.; Pescatore, M. Method for measuring the logarithm of the octanol–water partition coefficient by using short octadecyl–poly(vinyl alcohol) high-performance liquid chromatography columns. J. Chromatogr. A 2002, 952, 47–61. [Google Scholar] [CrossRef]
- Schmitt, M.; Greten, F. The inflammatory pathogenesis of colorectal cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Cho, K.-J.; Seo, J.-M.; Kim, J.-H. Bioactive lipoxygenase metabolites stimulation of NADPH oxidases and reactive oxygen species. Mol. Cells 2011, 32, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisser, H.; Göbel, T.; Krishnathas, G.M.; Kreiß, M.; Angioni, C.; Sürün, D.; Thomas, D.; Schmid, T.; Häfner, A.-K.; Kahnt, A. Knock-out of 5-lipoxygenase in overexpressing tumor cells-consequences on gene expression and cellular function. Cancer Gene Ther. 2023, 30, 108–123. [Google Scholar] [CrossRef]
- Agarwal, B.; Swaroop, P.; Protiva, P.; Raj, S.V.; Shirin, H.; Holt, P. COX-2 is needed but not sufficient for apoptosis induced by COX-2 selective inhibitors in colon cancer cells. Apoptosis 2003, 8, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191. [Google Scholar] [CrossRef] [Green Version]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, Ł.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Becker, E.D.; de Menezes, S.M.C.; Goodfellow, R.; Granger, P. NMR nomenclature. Nuclear spin properties and conventions for chemical shifts(IUPAC Recommendations 2001). Pure Appl. Chem. 2001, 73, 1795–1818. [Google Scholar]
- Liu, G.; Xu, B. Hydrogen bond donor solvents enabled metal and halogen-free Friedel–Crafts acylations with virtually no waste stream. Tetrahedron Lett. 2018, 59, 869–872. [Google Scholar] [CrossRef]
- Goto, T.; Ohta, K.; Suzuki, T.; Ohta, S.; Endo, Y. Design and synthesis of novel androgen receptor antagonists with sterically bulky icosahedral carboranes. Bioorg. Med. Chem. 2005, 13, 6414–6424. [Google Scholar] [CrossRef] [PubMed]
- Wodtke, R.; Wodtke, J.; Hauser, S.; Laube, M.; Bauer, D.; Rothe, R.; Neuber, C.; Pietsch, M.; Kopka, K.; Pietzsch, J.; et al. Development of an 18F-Labeled Irreversible Inhibitor of Transglutaminase 2 as Radiometric Tool for Quantitative Expression Profiling in Cells and Tissues. J. Med. Chem. 2021, 64, 3462–3478. [Google Scholar] [CrossRef] [PubMed]
- Brungs, M.; Rådmark, O.; Samuelsson, B.; Steinhilber, D. Sequential induction of 5-lipoxygenase gene expression and activity in Mono Mac 6 cells by transforming growth factor beta and 1,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1995, 92, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Kempińska, D.; Chmiel, T.; Kot-Wasik, A.; Mróz, A.; Mazerska, Z.; Namieśnik, J. State of the art and prospects of methods for determination of lipophilicity of chemical compounds. Trends Anal. Chem. 2019, 113, 54–73. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro Software System; Rigaku Corporation: Wroclaw, Poland, 2006. [Google Scholar]
- SCALE3 ABSPACK. Empirical Absorption Correction Using Spherical Harmonics; Oxford Diffraction: Abingdon, UK, 2006. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Putz, H.; Brandenburg, K. DIAMOND: Crystal and Molecular Structure Visualization; Crystal Impact GbR: Bonn, Germany, 2022. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | % Inhibition at 100 µM (A,B) | logD7.4, HPLC | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| R-830 | 93 | 85 | 3.91 |
| R-830-Cb | 18 | 6 | 2.37 |
| KME-4 | 64 | 97 | 3.79 |
| KME-4-Cb | 26 | n.i. | 1.87 |
| E-5110 | 66 | 105 | 3.35 |
| E-5110-Cb | n.i. | n.i. | 1.50 |
| S-2474 | 38 | 86 | 3.75 |
| S-2474-Cb | n.i. | n.i. | 1.88 |
| Compound | IC50 [µM] (A,B) | Compound | IC50 [µM] |
|---|---|---|---|
| R-830-Cb | 0.65 | R-830 | 0.26 |
| KME-4-Cb | 0.07 | KME-4 | 0.15 |
| E-5110-Cb | 0.22 | E-5110 | 0.12 |
| S-2474-Cb | <0.03 | S-2474 | <0.03 |
| Compounds | Assays | A375 | A549 | HCT116 | HT-29 | MDA-MB-231 |
|---|---|---|---|---|---|---|
| R-830 * | MTT | 16.7 ± 3.1 | 19.0 ± 0.7 | 20.6 ± 0.4 | 19.4 ± 0.4 | 33.3 ± 1.2 |
| CV | 29.4 ± 2.3 | 32.6 ± 1.3 | 30.8 ± 3.2 | 24.9 ± 1.3 | 34.0 ± 1.6 | |
| R-830-Cb | MTT | 35.9 ± 0.8 | 66.5 ± 1.7 | 40.9 ± 3.1 | 65.6 ± 5.3 | 46.5 ± 0.1 |
| CV | 47.2 ± 0.7 | 77.3 ± 1.7 | 49.1 ± 6 | 73.3 ± 3.4 | 55.6 ± 0.7 | |
| KME-4 * | MTT | 13.8 ± 0.6 | 21.9 ± 0.7 | 18.4 ± 4.6 | 18.5 ± 1.5 | 80.9 ± 7.3 |
| CV | 36.4 ± 1.3 | 36.3 ± 2.8 | 36.2 ± 1.9 | 36.2 ± 1.9 | 88.6 ± 2.4 | |
| KME-4-Cb | MTT | 83.3 ± 2.3 | 119.7 ± 7.0 | 88.5 ± 4.3 | 139.3 ± 5.4 | 119.7 ± 3.9 |
| CV | 155.2 ± 5.8 | 157.8 ± 3.0 | 127.0 ± 8.0 | 164.7 ± 8.5 | 132.7 ± 4.1 | |
| E-5110 * | MTT | 12.9 ± 1.1 | 22.1 ± 1.6 | 25.1 ± 1.1 | 25.6 ± 2.6 | 34.2 ± 1.2 |
| CV | 33.5 ± 1.7 | 41.1 ± 4.3 | 39.6 ± 2.8 | 39.8 ± 2.1 | 47.3 ± 1.7 | |
| E-5110-Cb | MTT | 69.3 ± 1.7 | 176.6 ± 14.5 | 143.9 ± 10.5 | 119.9 ± 9.7 | 200 ± 0.0 |
| CV | 103.2 ± 10.9 | 197.7 ± 5.7 | 193.1 ± 3.0 | 155.4 ± 6.0 | >200 | |
| S-2474 * | MTT | 13.2 ± 2.7 | 16.2 ± 1.2 | 17.5 ± 1.0 | 16.1 ± 1.1 | 34.8 ± 1.7 |
| CV | 19.2 ± 0.7 | 17.9 ± 1.2 | 23.7 ± 1.9 | 23.8 ± 1.7 | 43.5 ± 1.8 | |
| S-2474-Cb | MTT | 106.0 ± 6.3 | 121.8 ± 10.4 | 108.1 ± 11.5 | 81.8 ± 6.1 | >200 |
| CV | 135.5 ± 6.3 | 149.7 ± 4.3 | 133.4 ± 7.1 | 137.5 ± 7.8 | >200 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, S.; Jelača, S.; Laube, M.; George, S.; Hofmann, B.; Lönnecke, P.; Steinhilber, D.; Pietzsch, J.; Mijatović, S.; Maksimović-Ivanić, D.; et al. Synthesis and In Vitro Biological Evaluation of p-Carborane-Based Di-tert-butylphenol Analogs. Molecules 2023, 28, 4547. https://doi.org/10.3390/molecules28114547
Braun S, Jelača S, Laube M, George S, Hofmann B, Lönnecke P, Steinhilber D, Pietzsch J, Mijatović S, Maksimović-Ivanić D, et al. Synthesis and In Vitro Biological Evaluation of p-Carborane-Based Di-tert-butylphenol Analogs. Molecules. 2023; 28(11):4547. https://doi.org/10.3390/molecules28114547
Chicago/Turabian StyleBraun, Sebastian, Sanja Jelača, Markus Laube, Sven George, Bettina Hofmann, Peter Lönnecke, Dieter Steinhilber, Jens Pietzsch, Sanja Mijatović, Danijela Maksimović-Ivanić, and et al. 2023. "Synthesis and In Vitro Biological Evaluation of p-Carborane-Based Di-tert-butylphenol Analogs" Molecules 28, no. 11: 4547. https://doi.org/10.3390/molecules28114547