
Tetrasubstituted Pyrrole Derivative Mimetics of Protein–Protein Interaction Hot-Spot Residues: A Promising Class of Anticancer Agents Targeting Melanoma Cells
 ,
,  , ,
, ,  ,
,  ,
,
Abstract
:
1. Introduction
2. Results and Discussion
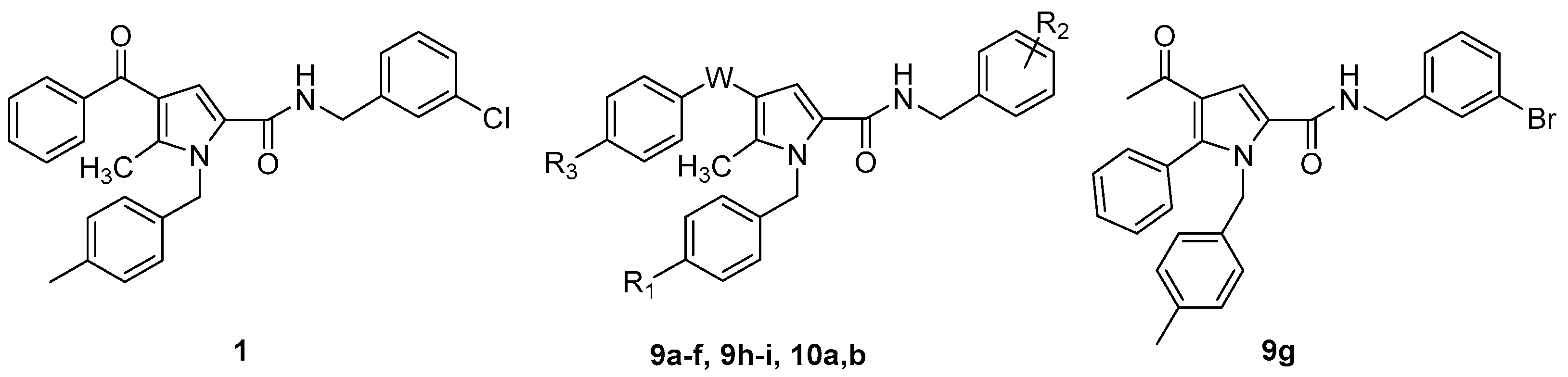
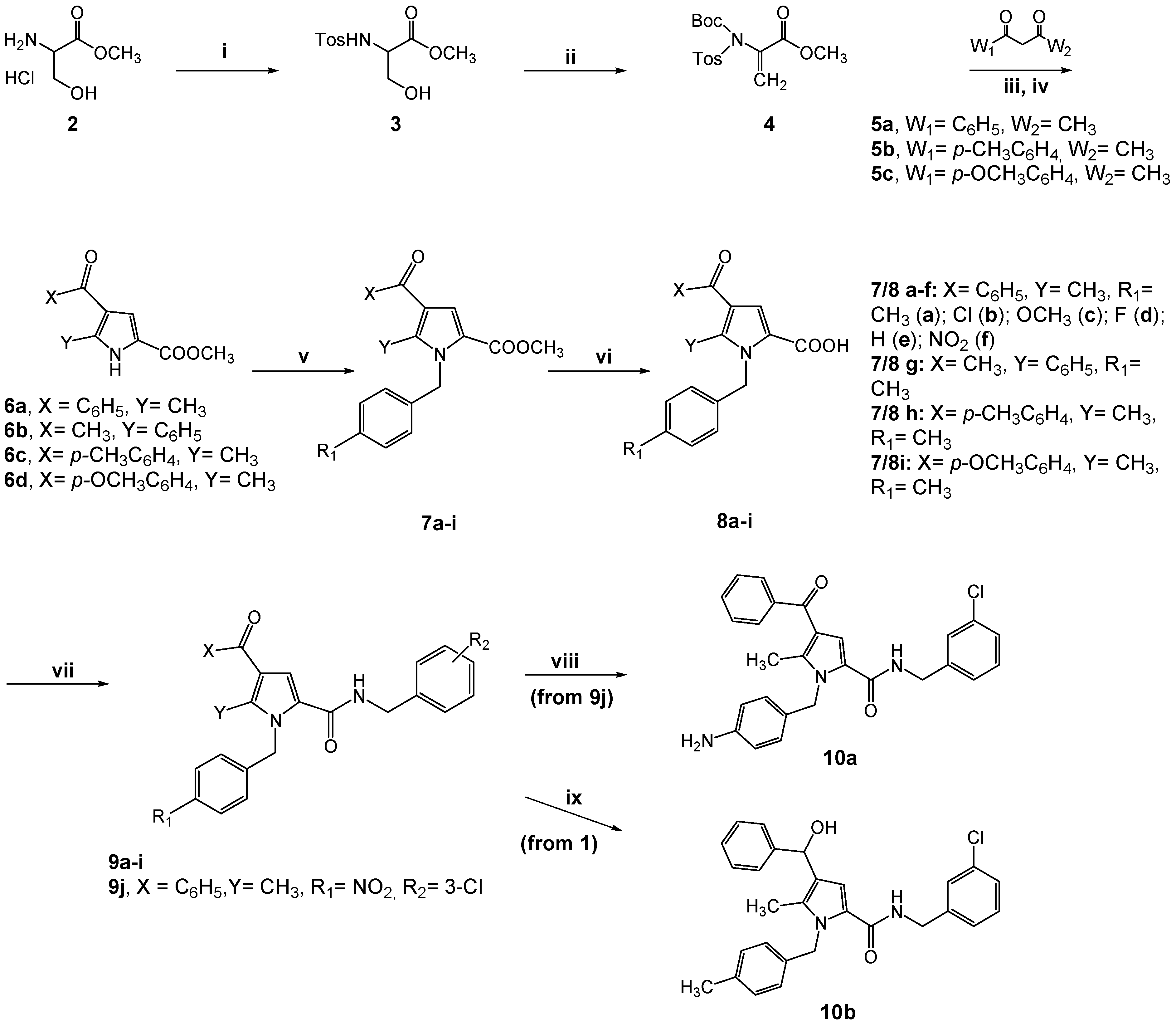
2.1. Design and Synthesis of the New TSP Derivatives
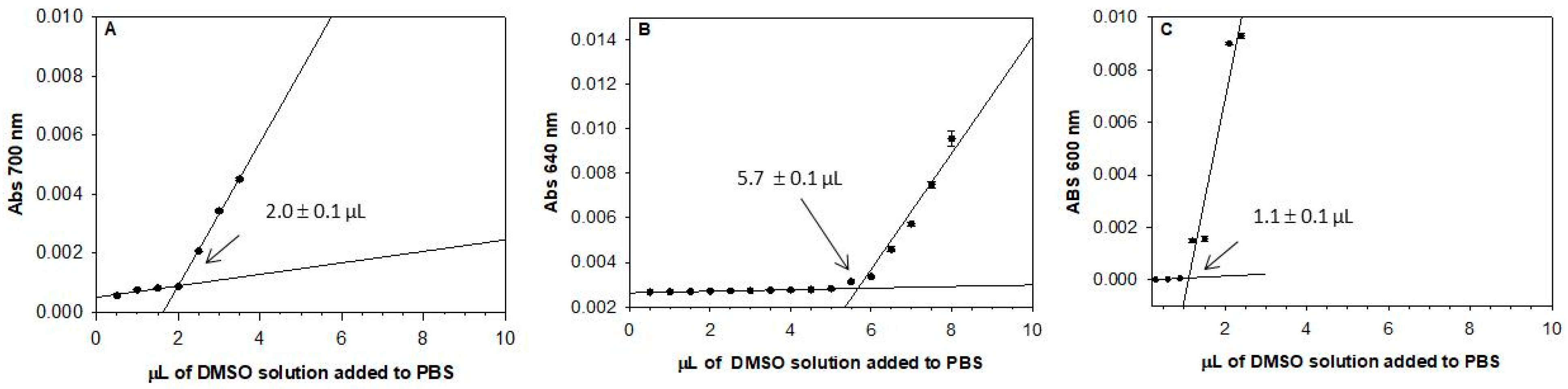
2.2. Solubility Assay
2.3. Biological Evaluation
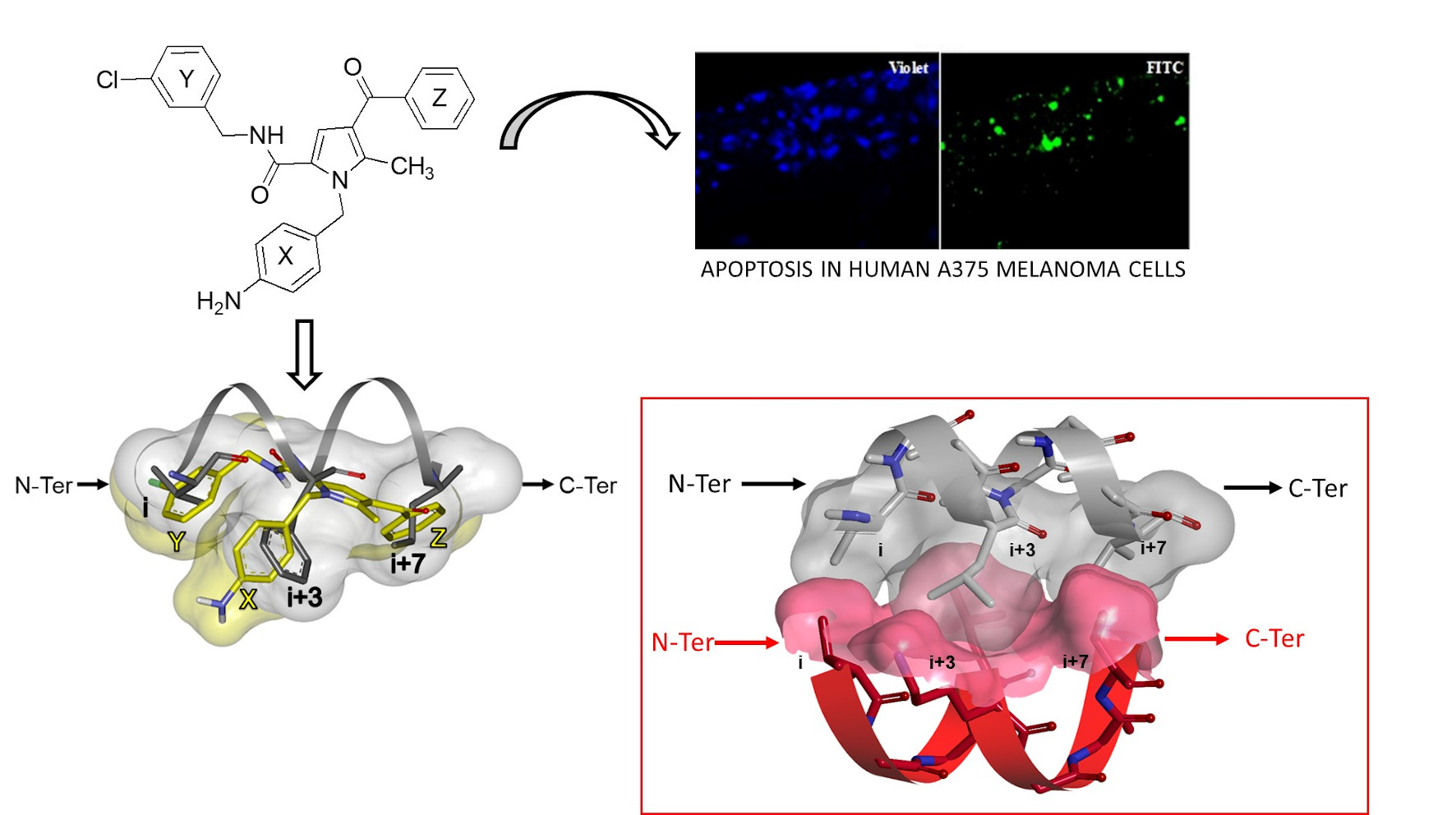
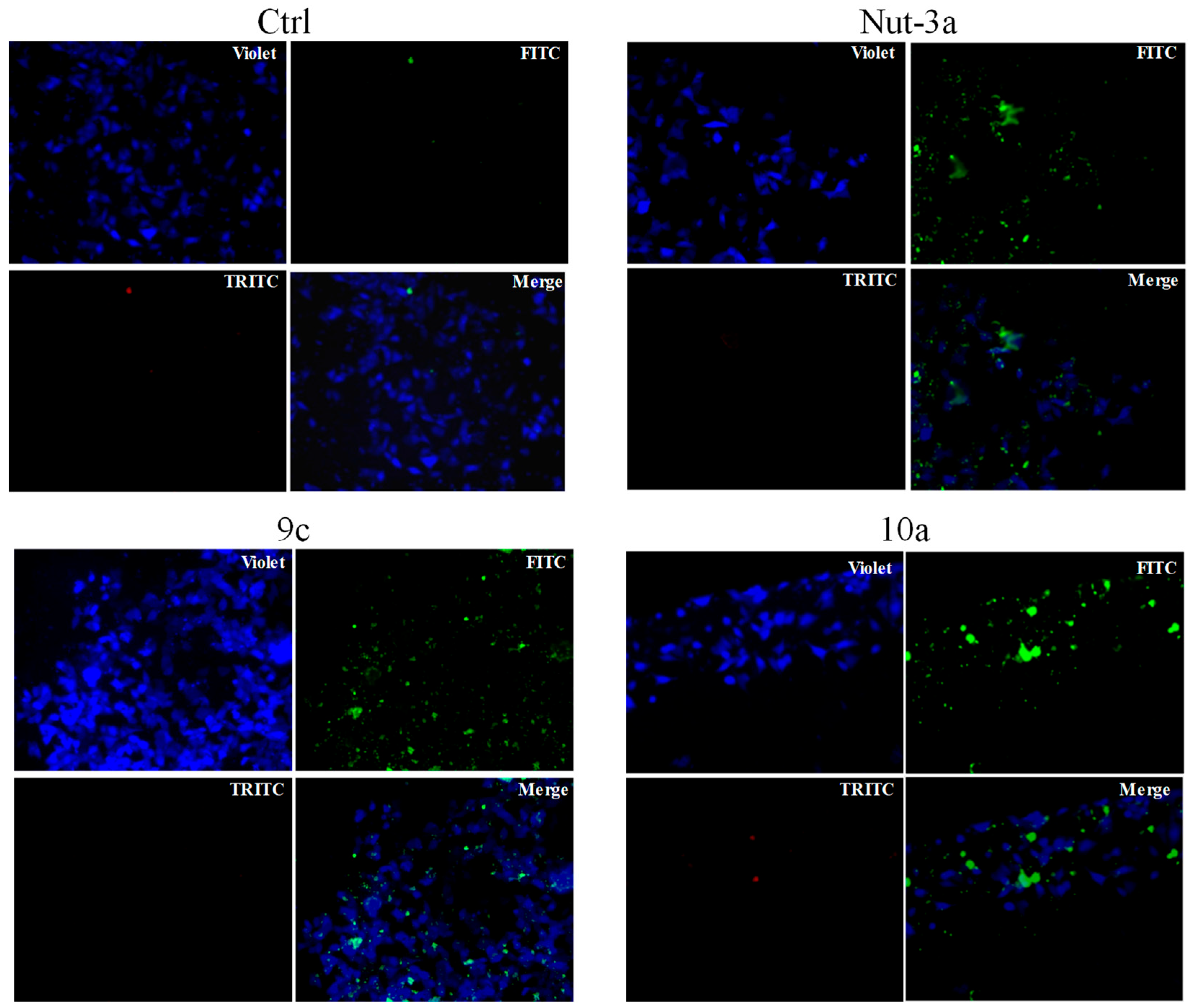
2.4. Apoptosis Studies
2.5. Computational Studies
2.5.1. Conformational Analysis
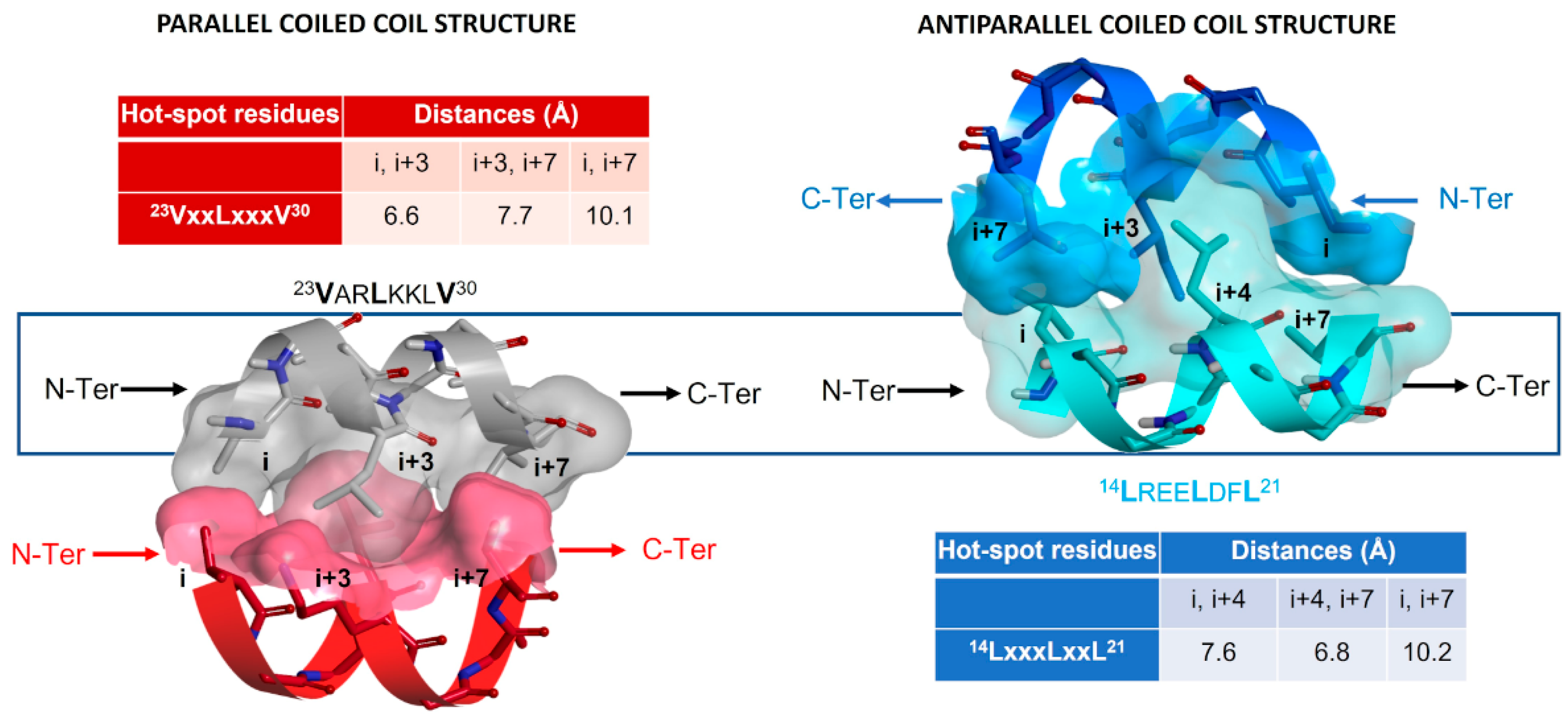
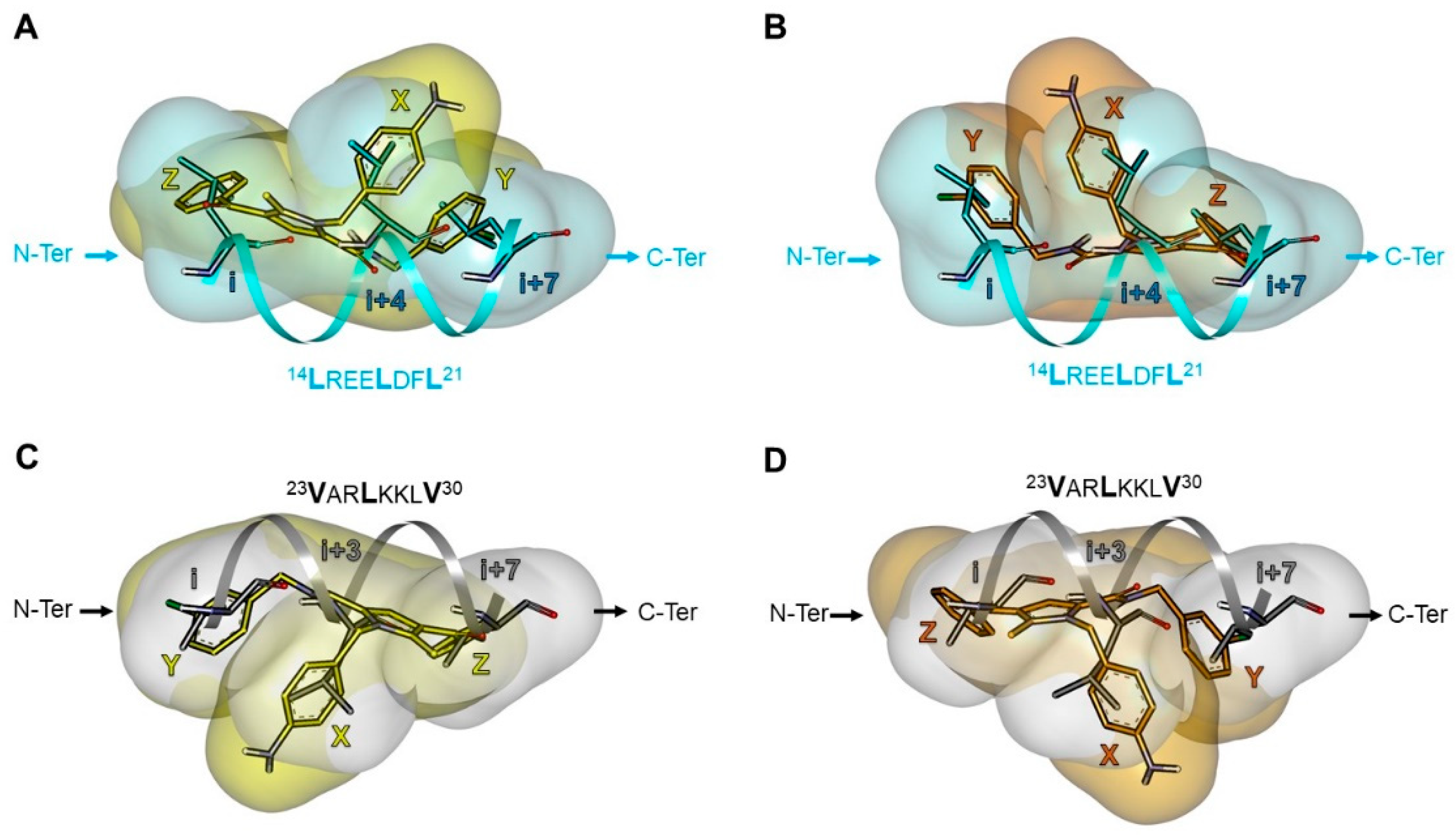
2.5.2. Investigation of the Peptidomimetic Ability of the New TSPs
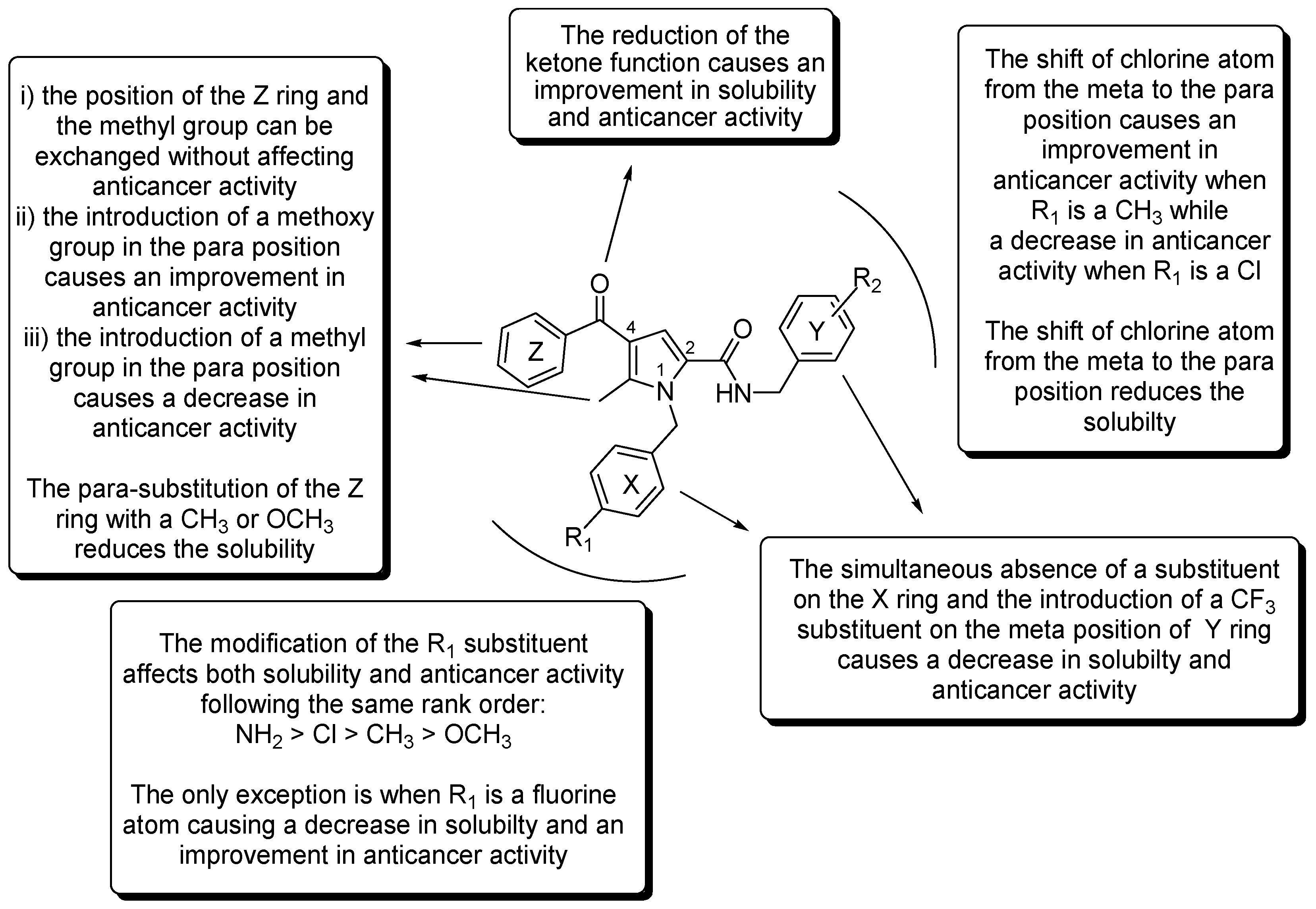
2.6. SAR Analysis
3. Materials and Methods
3.1. Chemistry
3.2. Cell Cultures
3.3. Bioscreens In Vitro for Anticancer Activity
3.4. Apoptosis/Necrosis Detection
3.5. Statistical Analysis
3.6. Solubility Assay
3.7. Molecular Modeling Studies
3.7.1. Conformational Analysis
3.7.2. Structural and Bioinformatics Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ivanov, A.A.; Khuri, F.R.; Fu, H. Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol. Sci. 2013, 34, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Bullock, B.N.; Jochim, A.L.; Arora, P.S. Assessing helical protein interfaces for inhibitor design. J. Am. Chem. Soc. 2011, 133, 14220–14223. [Google Scholar] [CrossRef] [PubMed]
- Algar, S.; Martín-Martínez, M.; González-Muñiz, R. Evolution in non-peptide α-helix mimetics on the road to effective protein-protein interaction modulators. Eur. J. Med. Chem. 2021, 211, 113015. [Google Scholar] [CrossRef] [PubMed]
- Ravindranathan, P.; Lee, T.-K.; Yang, L.; Centenera, M.M.; Butler, L.; Tilley, W.D.; Hsieh, J.-T.; Ahn, J.-M.; Raj, G.V. Peptidomimetic targeting of critical androgen receptor–coregulator interactions in prostate cancer. Nat. Commun. 2013, 4, 1923. [Google Scholar] [CrossRef] [PubMed]
- Becerril, J.; Hamilton, A.D. Helix mimetics as inhibitors of the interaction of the estrogen receptor with coactivator peptides. Angew. Chem. Int. Ed. 2007, 46, 4471–4473. [Google Scholar] [CrossRef]
- Kutzki, O.; Park, H.S.; Ernst, J.T.; Orner, B.P.; Yin, H.; Hamilton, A.D. Development of a potent Bcl-xL antagonist based on α-helix mimicry. J. Am. Chem. Soc. 2002, 124, 11838–11839. [Google Scholar] [CrossRef]
- Hoggard, L.R.; Zhang, Y.; Zhang, M.; Panic, V.; Wisniewski, J.A.; Ji, H. Rational design of selective small-molecule inhibitors for β-Catenin/B-cell lymphoma 9 protein–protein Interactions. J. Am. Chem. Soc. 2015, 137, 12249–12260. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; LaFrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef]
- Ribas, J.; Cubero, E.; Luque, F.J.; Orozco, M. Theoretical study of alkyl-pi and aryl-pi interactions. Reconciling theory and experiment. J. Org. Chem. 2002, 67, 7057–7065. [Google Scholar] [CrossRef]
- Persico, M.; Ramunno, A.; Maglio, V.; Franceschelli, S.; Esposito, C.; Carotenuto, A.; Brancaccio, D.; De Pasquale, V.; Pavone, L.M.; Varra, M.; et al. New anticancer agents mimicking protein recognition motifs. J. Med. Chem. 2013, 56, 6666–6680. [Google Scholar] [CrossRef]
- Proto, M.C.; Fiore, D.; Forte, G.; Cuozzo, P.; Ramunno, A.; Fattorusso, C.; Gazzerro, P.; Pascale, M.; Franceschelli, S. Tetra-substituted pyrrole derivatives act as potent activators of p53 in melanoma Cclls. Investig. New Drugs 2020, 38, 634–649. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Xie, W.-H.; Shiu, W.-Y.; Mackay, D. A Review of the effect of salts on the solubility of organic compounds in seawater. Mar. Environ. Res. 1997, 44, 429–444. [Google Scholar] [CrossRef]
- Zangi, R.; Hagen, M.; Berne, B.J. Effect of ions on the hydrophobic interaction between two plates. J. Am. Chem. Soc. 2007, 129, 4678–4686. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Budyka, M.F.; Oshkin, I.V. Comparative semiempirical and DFT study of styrylnaphthalenes and styrylquinolines and their photocyclization products. Int. J. Quantum Chem. 2011, 111, 3673–3680. [Google Scholar] [CrossRef]
- Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J. Comp. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef]
- DiGiammarino, E.L.; Filippov, I.; Weber, J.D.; Bothner, B.; Kriwacki, R.W. Solution structure of the p53 regulatory domain of the p19Arf tumor suppressor protein. Biochemistry 2001, 40, 2379–2386. [Google Scholar] [CrossRef]
- Irace, C.; Misso, G.; Capuozzo, A.; Piccolo, M.; Riccardi, C.; Luchini, A.; Caraglia, M.; Paduano, L.; Montesarchio, D.; Santamaria, R. Antiproliferative effects of ruthenium-based nucleolipidic nanoaggregates in human models of breast cancer in vitro: Insights into their mode of action. Sci. Rep. 2017, 7, 45236. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.E.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field (CGenFF): A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, R. Unconstrained optimization. In Practical Methods of Optimization; John Wiley & Sons: New York, NY, USA, 1980; Volume 1. [Google Scholar]
- Nakamura, N.; Michinori, O. Restricted Rotation in Aromatic Ketones. I. Substituent Effects on the Barrier to Rotation about the Benzene-to-Carbonyl Bond. Bull. Chem. Soc. Jpn 1972, 45, 2565–2570. [Google Scholar] [CrossRef]
- Olsen, R.A.; Liu, L.; Ghaderi, N.; Johns, A.; Hatcher, M.E.; Mueller, L.J. The amide rotational barriers in picolinamide and nicotinamide: NMR and ab initio studies. J. Am. Chem. Soc. 2003, 125, 10125–10132. [Google Scholar] [CrossRef]
- Still, W.C.; Tempczyk, A.; Hawley, R.C.; Hendrickson, T. Semianalytical treatment of solvation for molecular mechanics and dynamics. J. Am. Chem. Soc. 1990, 112, 6127–6129. [Google Scholar] [CrossRef]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; McNaught, A.D., Wilkinson, A., Eds.; the “Gold Book”; Online Version (2019-) Created by Chalk, S.J.; Blackwell Scientific Publications: Oxford, UK, 1997; ISBN 0-9678550-9-8. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple sequence and structure alignment. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
| Cpd | W | R1 | R2 | R3 |
| 1 | C=O | CH3 | 3-Cl | H |
| 9a | C=O | Cl | 3-Cl | H |
| 9b | C=O | OCH3 | 3-Cl | H |
| 9c | C=O | F | 3-Cl | H |
| 9d | C=O | CH3 | 4-Cl | H |
| 9e | C=O | Cl | 4-Cl | H |
| 9f | C=O | H | 3-CF3 | H |
| 9h | C=O | CH3 | 3-Cl | CH3 |
| 9i | C=O | CH3 | 3-Cl | OCH3 |
| 10a | C=O | NH2 | 3-Cl | H |
| (±)10b | CHOH | CH3 | 3-Cl | H |
| Cpd | Solubility a (µM) |
|---|---|
| 1 | 3.5 ± 0.1 |
| 9a | 3.9 ± 0.1 |
| 9b | 3.1 ± 0.2 |
| 9c | 3.9 ± 0.2 |
| 9d | 3.1 ± 0.2 |
| 9e | 3.5 ± 0.3 |
| 9f | 2.6 ± 0.1 |
| 9g | 1.5 ± 0.2 |
| 9h | <1.0 |
| 9i | <1.0 |
| 10a | 10.5 ± 0.1 |
| (±)10b | 6.7 ± 0.6 |
| nutlin-3a | 2.4 ± 0.2 |
| Cpd | A375 a IC50 (µM) b | L6 c IC50 (µM) b | SI d | HCT-116 e IC50 (µM) b |
|---|---|---|---|---|
| 1 | 20 ± 4 | 42 ± 3 | 2.1 | 41.4 ± 6 |
| 9a | 18 ± 4 | 48 ± 5 | 2.7 | 36.4 ± 8 |
| 9b | 24 ± 14 | 40 ± 5 | 1.7 | 46 ± 18 |
| 9c | 13 ± 4 | 35 ± 4 | 2.7 | 55.9 ± 7 |
| 9d | 15 ± 5 | 25 ± 6 | 1.7 | 35.4 ± 6 |
| 9e | 23 ± 6 | 38 ± 2 | 1.6 | 34.6 ± 6 |
| 9f | 27 ± 8 | 35 ± 7 | 1.3 | 28.4 ± 7 |
| 9g | 18 ± 5 | 71 ± 5 | 3.9 | 34 ± 6 |
| 9h | 24 ± 6 | 42 ± 7 | 1.7 | 56.3 ± 15 |
| 9i | 16 ± 6 | 66 ± 5 | 4.2 | 167.6 ± 9 |
| 10a | 10 ± 4 | 55 ± 7 | 5.6 | 41.3 ± 6 |
| (±)10b | 15 ± 5 | 54 ± 6 | 3.6 | 25 ± 5 |
| nutlin-3a | 15 ± 3 | 18 ± 4 | 1.2 | 12.5 ± 5 |
 | ||||||||||
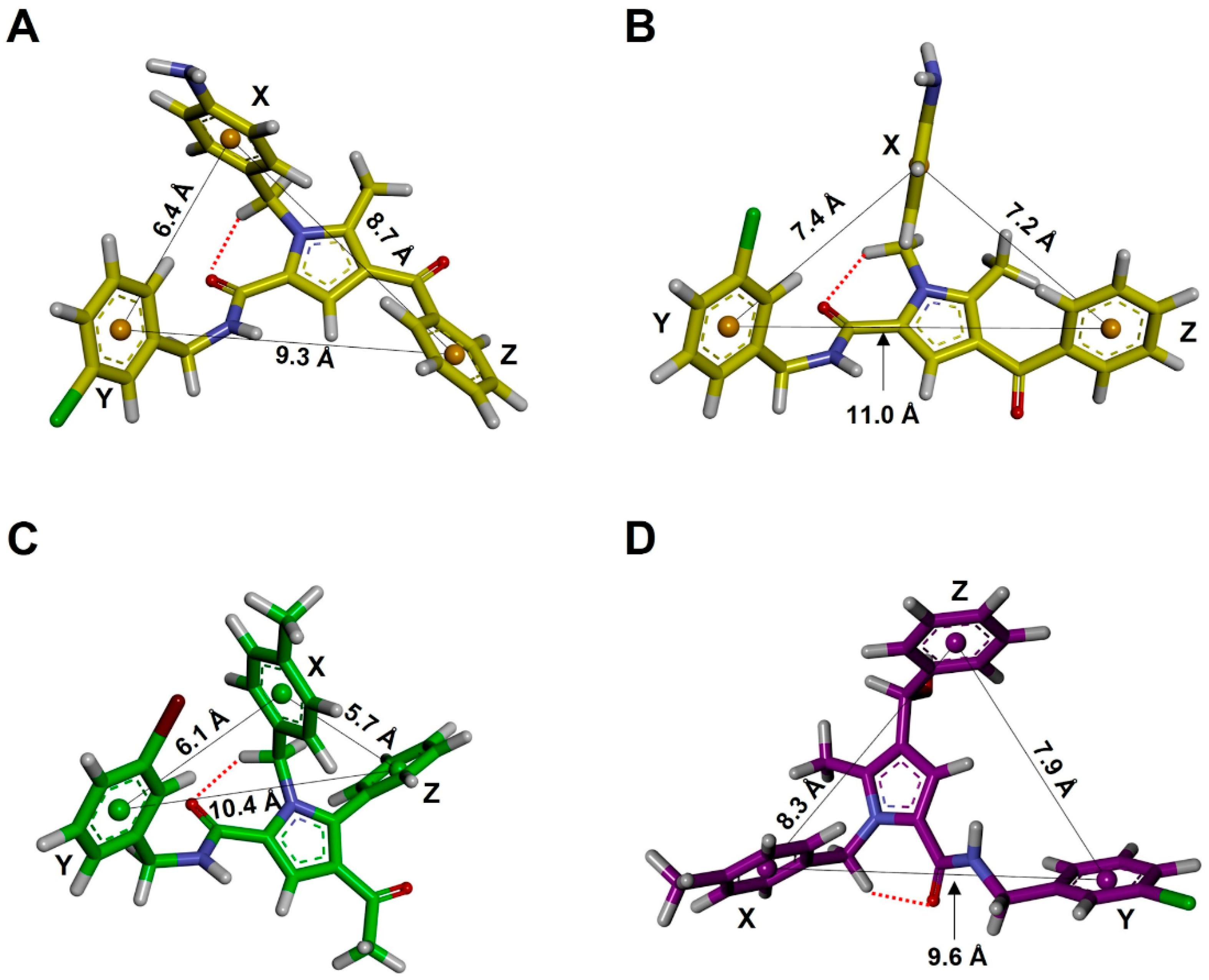
| Family a | Sub-Family | ΔEGM (kcal/mol) | Distances (Å) | Torsional Angles (°) b–f | ||||||
| d1 (X-Y) | d2 (X-Z) | d3 (Y-Z) | τ1 | τ2 | τ3 | τ4 | τ5 | |||
| TCC | I | 0.00 | 6.4 | 8.7 | 9.3 | −174 | 26 | −21 | 77 | −112 |
| TCC | II | 0.49 | 5.6 | 9.3 | 10.2 | −174 | 28 | 22 | 75 | −82 |
| TCT | I | 1.57 | 7.4 | 7.2 | 11.0 | −174 | 27 | −148 | 102 | −111 |
| TCT | III | 1.65 | 9.7 | 7.2 | 10.7 | −173 | 27 | −148 | 101 | 123 |
| Pharmacophore | TSP Ring | PPI Motif | |||
|---|---|---|---|---|---|
| 23VxxLxxxV30 | 14LxxxLxxL21 | ||||
| Hot-Spot Residues a | |||||
| X | i + 3 | i + 3 | i + 4 | i + 4 | |
| 1 | Y | i | i + 7 | i | i + 7 |
| Z | i + 7 | i | i + 7 | i | |
| X | i + 3 | i + 3 | i + 4 | i + 4 | |
| 2 | Y | i | i + 7 | i | i + 7 |
| Z | i + 7 | i | i + 7 | i | |
| X | i + 3 | i + 3 | i + 4 | i + 4 | |
| 3 | Y | i | i + 7 | i | i + 7 |
| Z | i + 7 | i | i + 7 | i | |
| X | i | i + 7 | i | i + 7 | |
| 4 | Y | i + 7 | i | i + 7 | i |
| Z | i + 3 | i + 3 | i + 4 | i + 4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Persico, M.; Galatello, P.; Ferraro, M.G.; Irace, C.; Piccolo, M.; Abduvakhidov, A.; Tkachuk, O.; d’Aulisio Garigliota, M.L.; Campiglia, P.; Iannece, P.; et al. Tetrasubstituted Pyrrole Derivative Mimetics of Protein–Protein Interaction Hot-Spot Residues: A Promising Class of Anticancer Agents Targeting Melanoma Cells. Molecules 2023, 28, 4161. https://doi.org/10.3390/molecules28104161
Persico M, Galatello P, Ferraro MG, Irace C, Piccolo M, Abduvakhidov A, Tkachuk O, d’Aulisio Garigliota ML, Campiglia P, Iannece P, et al. Tetrasubstituted Pyrrole Derivative Mimetics of Protein–Protein Interaction Hot-Spot Residues: A Promising Class of Anticancer Agents Targeting Melanoma Cells. Molecules. 2023; 28(10):4161. https://doi.org/10.3390/molecules28104161
Chicago/Turabian StylePersico, Marco, Paola Galatello, Maria Grazia Ferraro, Carlo Irace, Marialuisa Piccolo, Avazbek Abduvakhidov, Oleh Tkachuk, Maria Luisa d’Aulisio Garigliota, Pietro Campiglia, Patrizia Iannece, and et al. 2023. "Tetrasubstituted Pyrrole Derivative Mimetics of Protein–Protein Interaction Hot-Spot Residues: A Promising Class of Anticancer Agents Targeting Melanoma Cells" Molecules 28, no. 10: 4161. https://doi.org/10.3390/molecules28104161