
Nature of Luminescence and Pharmacological Activity of Sulfaguanidine

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
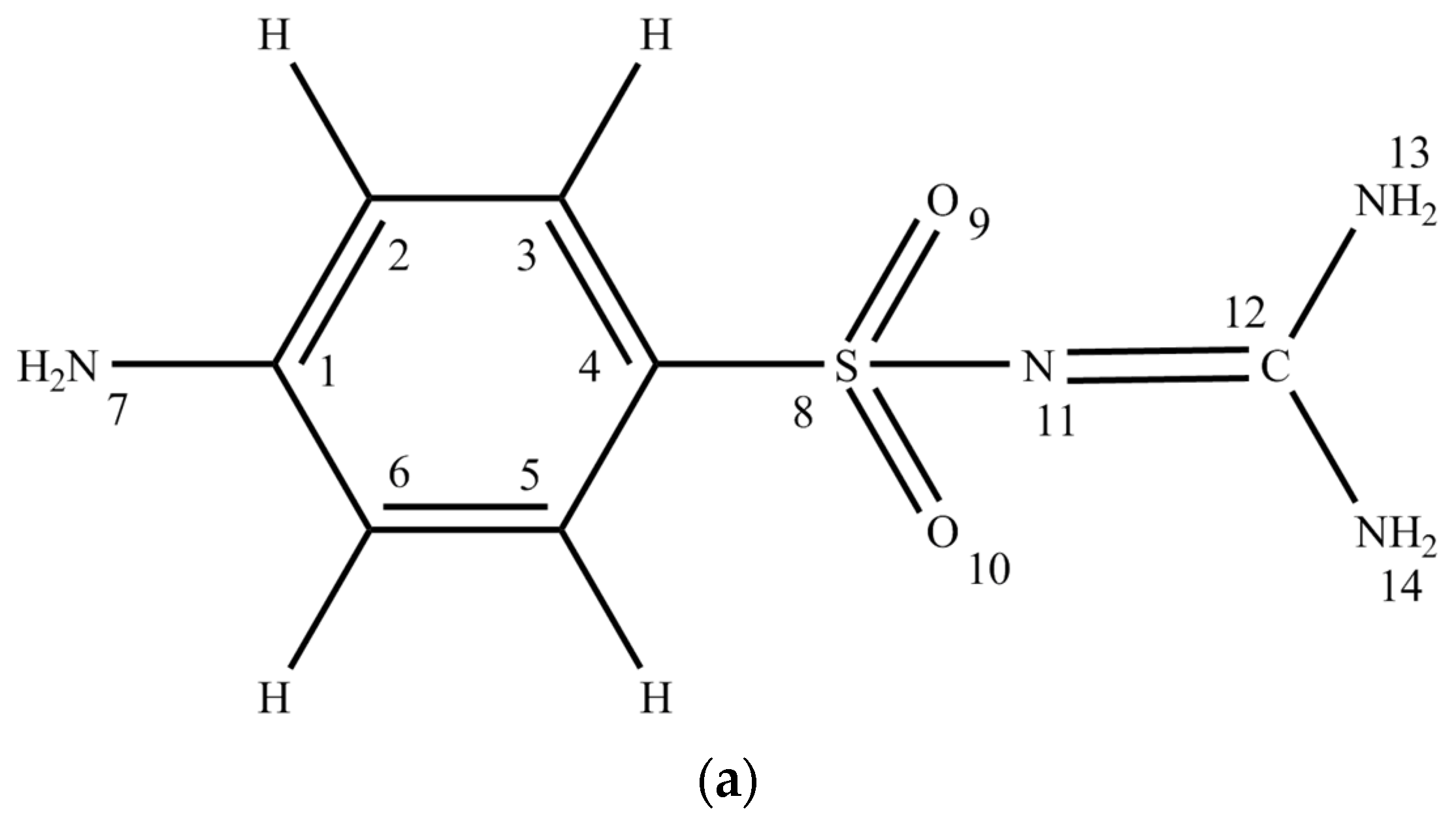
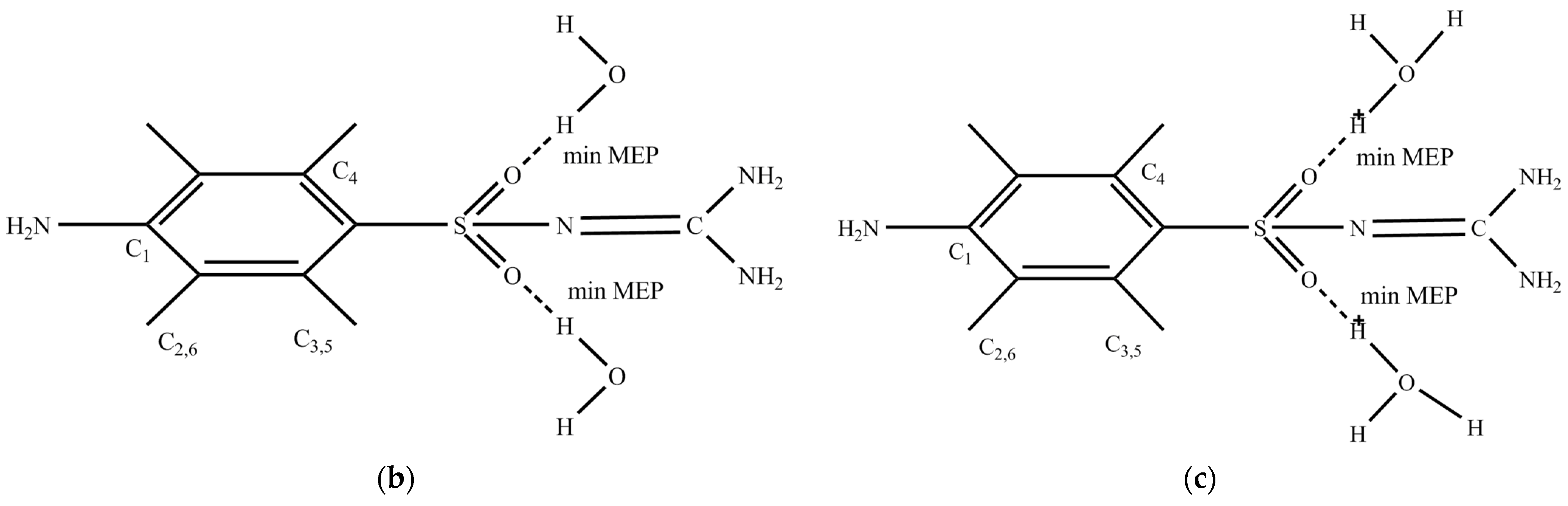
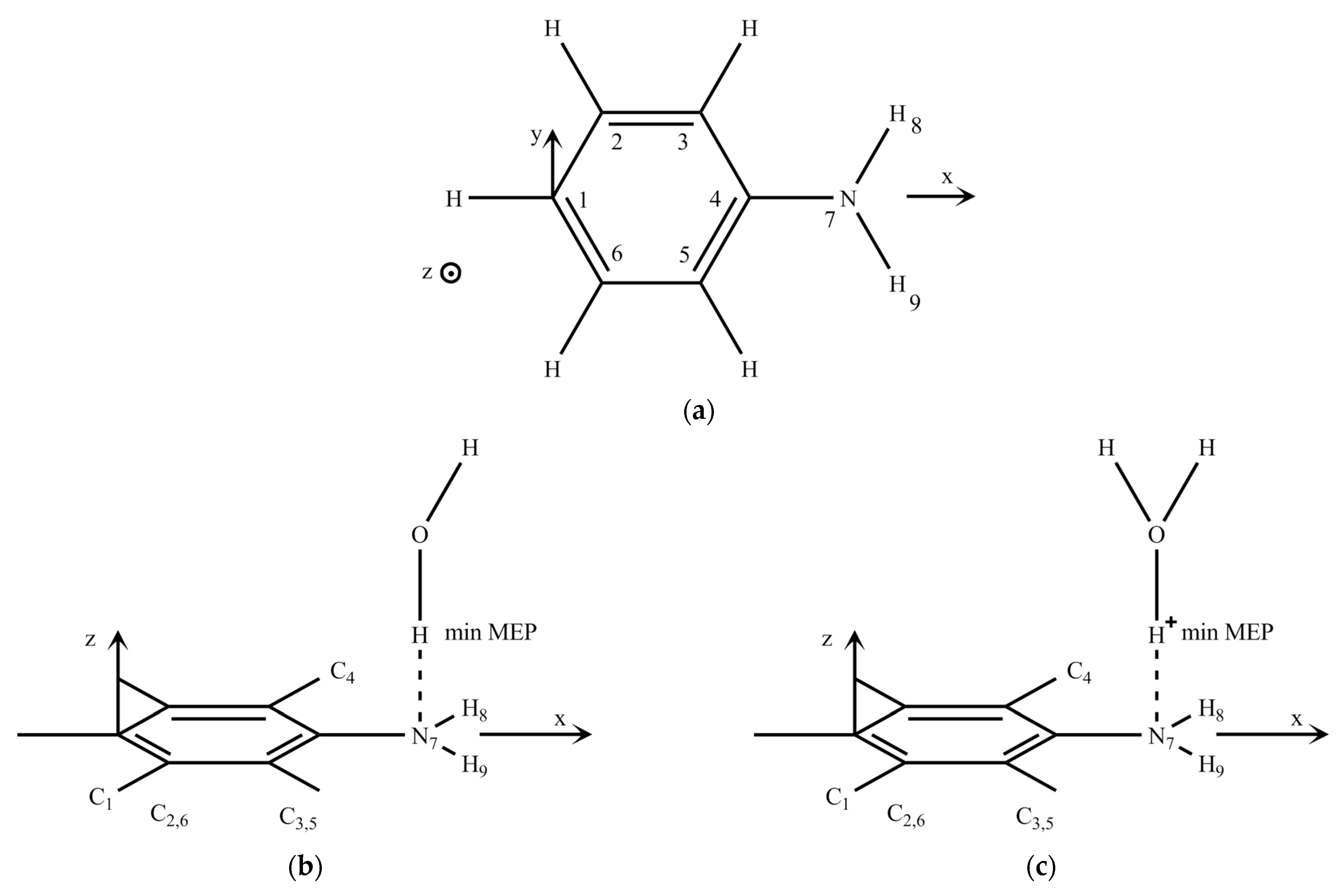
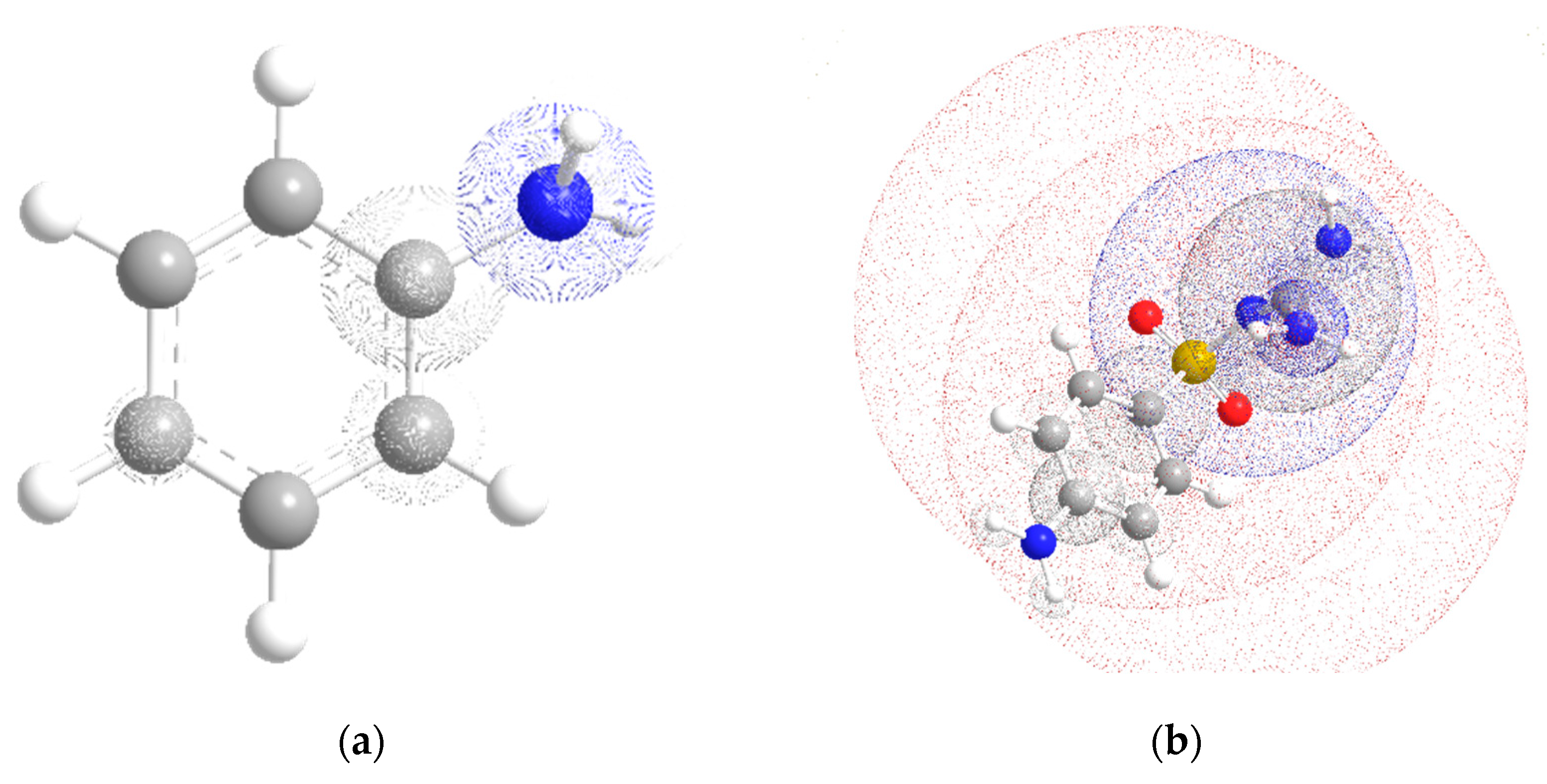
2.1. Conformational Design
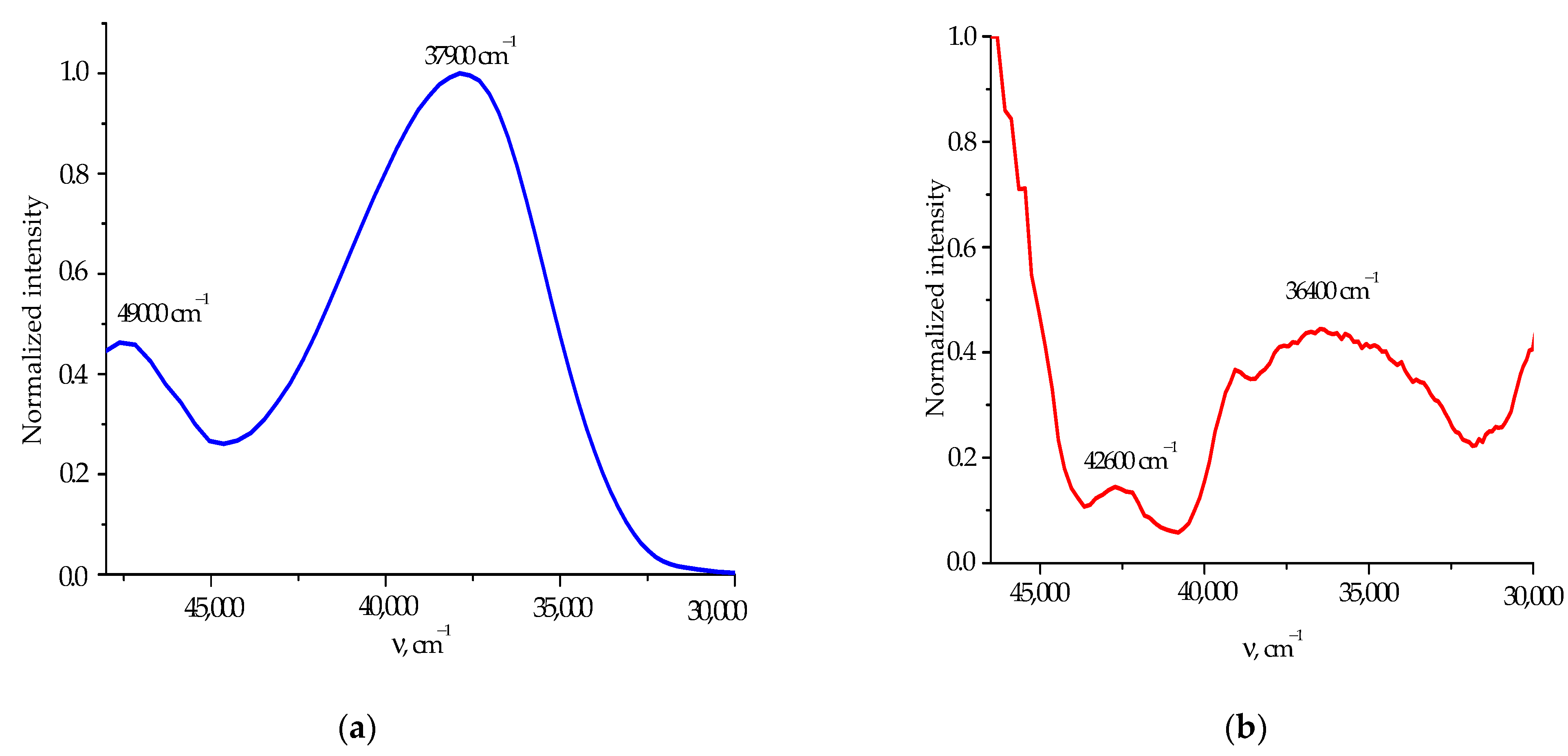
2.2. Discussion
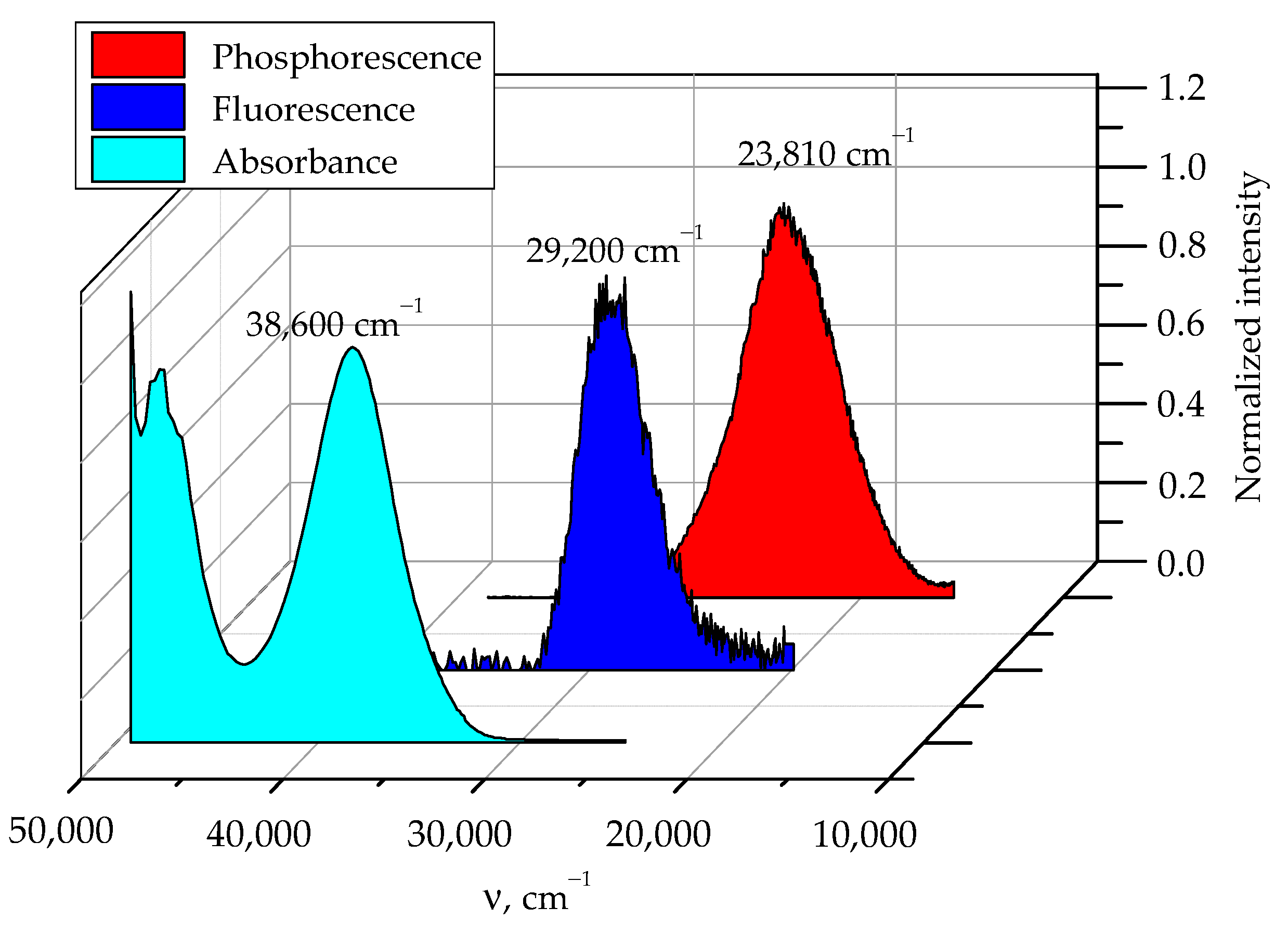
2.3. Fluorescence Quantum Yield of Sulfaguanidine
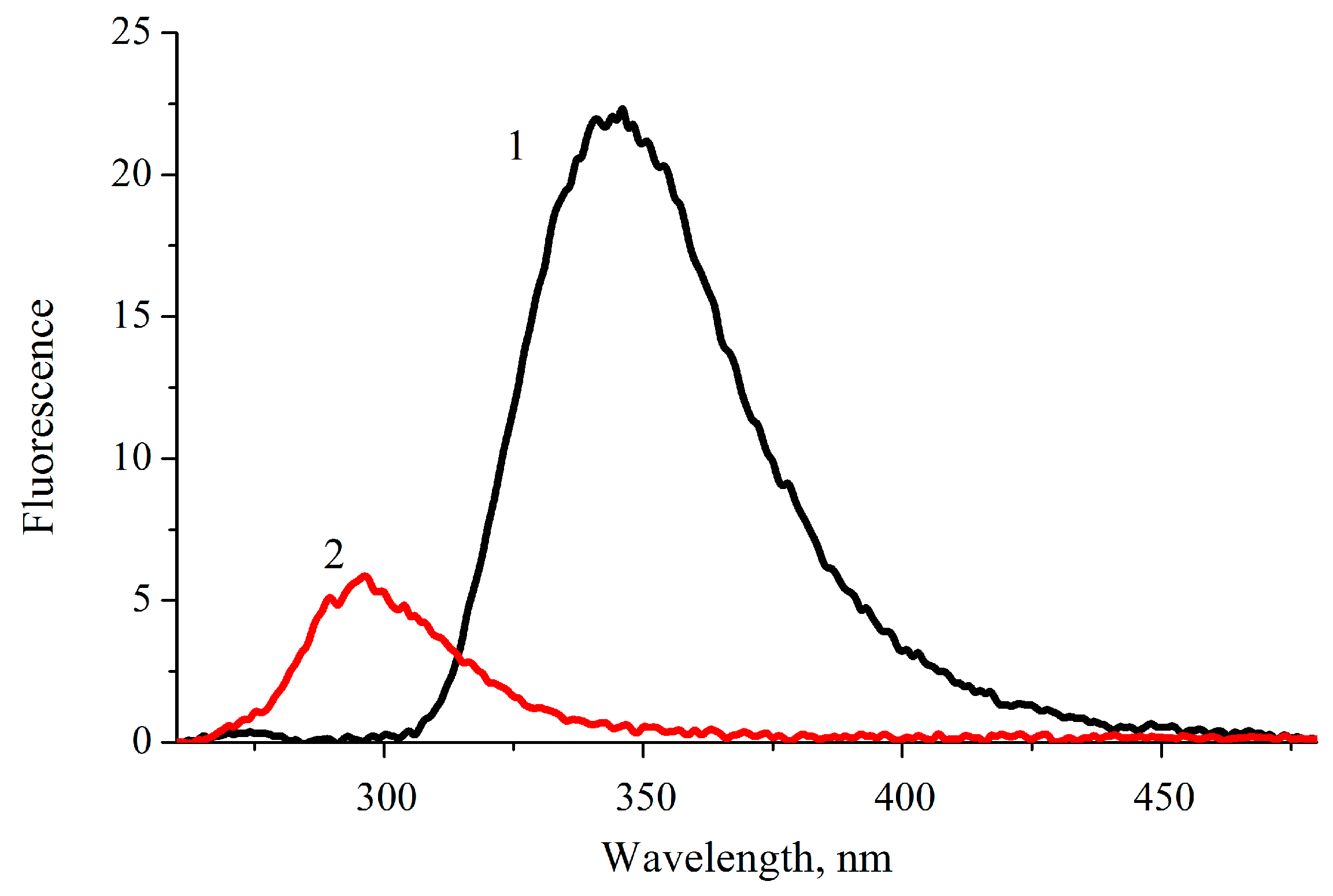
2.4. Phosphorescence of Sulfaguanidine in Water
3. Materials and Methods
3.1. Experimental and Object
3.2. Calculation of the Theoretical Quantum Yield Using Quantum Chemistry
3.3. Measurement of the Fluorescence Quantum Yield from a Standard
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chuang, K.V.; Gunsalus, L.M.; Keiser, M.J. Learning molecular representations for medicinal chemistry. J. Med. Chem. 2020, 63, 8705–8722. [Google Scholar] [CrossRef] [PubMed]
- Dardeer, H.M.; Toghan, A.; Zaki, M.E.A.; Elamary, R.B. Design, synthesis and evaluation of novel antimicrobial polymers based on the inclusion of polyethylene glycol/TiO2 nanocomposites in cyclodextrin as drug carriers for sulfaguanidine. Polymers 2022, 14, 227. [Google Scholar] [CrossRef] [PubMed]
- El Hassani, N.E.A.; Llobet, E.; Popescu, L.-M.; Ghita, M.; Bouchikhi, B.; Bari, N.E. Development of a highly sensitive and selective molecularly imprinted electrochemical sensor for sulfaguanidine detection in honey samples. J. Electroanal. Chem. 2018, 823, 647–655. [Google Scholar] [CrossRef]
- Priscillal, I.J.D.; Alothman, A.A.; Wang, S.-F.; Arumugam, R. Lanthanide type of cerium sulfide embedded carbon nitride composite modified electrode for potential electrochemical detection of sulfaguanidine. Microchim. Acta 2021, 188, 313. [Google Scholar] [CrossRef]
- Aouad, M.R.; Khan, D.J.O.; Said, M.A.; Al-Kaff, N.S.; Rezki, N.; Ali, A.A.; Bouqellah, N.; Hagar, M. Novel 1,2,3-triazole derivatives as potential inhibitors against Covid-19 main protease: Synthesis, characterization, molecular docking and DFT studies. Chem. Select. 2021, 6, 3468–3486. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.D.N.; Nagpal, S.; Sadqi, M.; Muñoz, V. A modular approach to map out the conformational landscapes of unbound intrinsically disordered proteins. Biophys. Comput. Biol. 2022, 119, e2113572119. [Google Scholar] [CrossRef] [PubMed]
- Ushakov, R.V.; Brodsky, S.E. Studying the possibilities of using antimicrobial chemotherapy and prevention in dental practice (organizational sociological study). Dentist 2007, 8, 2–6. [Google Scholar]
- Hu, J.; Li, X.; Liu, F.; Fu, W.; Lin, L.; Li, B. Comparison of chemical and biological degradation of sulfonamides: Solving the mystery of sulfonamide transformation. J. Hazard. Mater. 2022, 424, 127661. [Google Scholar] [CrossRef]
- Duan, W.; Cui, H.; Jia, X.; Huang, X. Occurrence and ecotoxicity of sulfonamides in the aquatic environment: A review. Sci. Total Environ. 2022, 820, 153178. [Google Scholar] [CrossRef]
- Alexandrov, A.A.; Bondarenko, I.Z.; Kukharenko, S.S.; Yadrikhinskaya, M.N.; Martyanova, I.I.; Solyanik, Y.A.; Drozdova, E.N.; Maiorov, A.Y. Diabetes mellitus and coronary heart disease: The search for solutions. Diabetes Mellit. 2005, 3, 34–38. [Google Scholar] [CrossRef]
- Irfan, A.; Ahmad, S.; Hussain, S.; Batool, F.; Riaz, H.; Zafar, R.; Kotwica-Mojzych, K.; Mojzych, M. Recent Updates on the Synthesis of Bioactive Quinoxaline-Containing Sulfonamides. Appl. Sci. 2021, 11, 5702. [Google Scholar] [CrossRef]
- Kokoszka, K.; Wilk, J.; Felis, E.; Bajkacz, S. Application of UHPLC-MS/MS method to study occurrence and fate of sulfonamide antibiotics and their transformation products in surface water in highly urbanized areas. Chemosphere 2021, 283, 131189. [Google Scholar] [CrossRef] [PubMed]
- Markushyna, Y.; Schüßlbauer, C.M.; Ullrich, T.; Guldi, D.M.; Antonietti, M.; Savateev, A. Chromoselective Synthesis of Sulfonyl Chlorides and Sulfonamides with Potassium Poly(heptazine imide) Photocatalyst. Angew. Chem. Int. Ed. 2021, 60, 20543–20550. [Google Scholar] [CrossRef]
- Aleksandrov, A.A.; Bondarenko, I.Z.; Kukharenko, S.S.; Yadrikhinskaya, M.N.; Vilkov, V.G.; Akhmatova, F.D.; Aleksandrova, V.K.; Artamonova, N.V.; Solovieva, O.E.; Dedov, I.I. Hypoglycemic sulfonamides and the “ischemic threshold” of the heart in patients with type 2 diabetes mellitus with coronary heart disease. Diabetes Mellit. 2002, 4, 18–22. [Google Scholar] [CrossRef]
- Taha, M.; Imran, S.; Salahuddin, M.; Iqbal, M.; Rahim, F.; Uddin, N.; Shehzad, A.; Farooq, R.K.; Alomari, M.; Khan, K.M. Evaluation and docking of indole sulfonamide as a potent inhibitor of α-glucosidase enzyme in streptozotocin –induced diabetic albino wistar rats. Bioorganic Chem. 2021, 110, 104808. [Google Scholar] [CrossRef] [PubMed]
- Zajda, A.; Sikora, J.; Huttunen, K.M.; Markowicz-Piasecka, M. Structural Comparison of Sulfonamide-Based Derivatives That Can Improve Anti-Coagulation Properties of Metformin. Int. J. Mol. Sci. 2021, 23, 4132. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Huttunen, K.M.; Mateusiak, L.; Mikiciuk-Olasik, E.; Sikora, J. Sulfenamide and sulfonamide derivatives of metformin can exert anticoagulant and profibrinolytic properties. Chem. -Biol. Interact. 2018, 284, 126–136. [Google Scholar] [CrossRef]
- Pashinyan, A.G. Therapy of urinary tract infections. Med. Counc. 2011, 3–4, 46–47. [Google Scholar]
- Rybakova, M.V.; Zhukov, S.V. A device with antibacterial and hemostatic properties for first aid for wounds. Int. Stud. Sci. Bull. 2017, 6, 19–24. [Google Scholar]
- Ingalls, J.W., Jr. The potential value of sulfaguanidine in urology. Science 1954, 119, 191–192. [Google Scholar] [CrossRef]
- Smyth, C.J.; Finkelstein, M.B.; Gould, S.E.; Koppa, T.M.; Leeder, F.S. Acute Bacillary Dysentery (Flexner): Treatment with Sulfaguanidine and Succinylsulfathiazole. J. Am. Med. Assoc. 1943, 121, 1325–1330. [Google Scholar] [CrossRef]
- Rose, F.L.; Spinks, A. The absorption of some sulphaguanidine derivatives in mice. Br. J. Pharmacol. Chemother. 1947, 2, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Duan, H.; Zou, J.; Cao, J.; Wan, C.; Zhang, C.; Ma, H. A DOPO Derivative Constructed by Sulfaguanidine and Thiophene toward Enhancing Fire Safety, Smoke Suppression, and Mechanical Properties of Epoxy Resin. Macromol. Mater. Eng. 2021, 306, 2100569. [Google Scholar] [CrossRef]
- Fatima, A.; Singh, M.; Singh, N.; Savita, S.; Verma, I.; Siddiqui, N.; Jave, S. Investigations on experimental, theoretical spectroscopic, electronic excitations, molecular docking of Sulfaguanidine (SG): An antibiotic drug. Chem. Phys. Lett. 2021, 783, 139049. [Google Scholar] [CrossRef]
- Lolak, N.; Akocak, S.; Durgun, M.; Duran, H.E.; Necip, A.; Türkeş, C.; Işık, M.; Beydemir, Ş. Novel bis-ureido-substituted sulfaguanidines and sulfisoxazoles as carbonic anhydrase and acetylcholinesterase inhibitors. Mol. Divers. 2022, 1–15. [Google Scholar] [CrossRef]
- Rezki, N.; Almehmadi, M.A.; Ihmaid, S.; Shehata, A.M.; Omar, A.M.; Ahme, H.E.A.; Aouad, M.R. Novel scaffold hopping of potent benzothiazole and isatin analogues linked to 1,2,3-triazole fragment that mimic quinazoline epidermal growth factor receptor inhibitors: Synthesis, antitumor and mechanistic analyses. Bioorganic Chem. 2020, 103, 104133. [Google Scholar] [CrossRef]
- Tarasov, G.P.; Khairullina, V.R.; Gerchikov, A.Y.; Zarudiy, F.S. Relationship “structure-property” in a series of inhibitors of the activity of a specific potassium channel hERG. Bull. Bashkir Univ. 2012, 17, 30–35. [Google Scholar]
- Uhlemann, T.; Berden, G.; Oomens, J. Preferred protonation site of a series of sulfa drugs in the gas phase revealed by IR spectroscopy. Eur. Phys. J. D 2021, 75, 21–34. [Google Scholar] [CrossRef]
- Mohammed, G.I.; El-Ghamry, H.A.; Saber, A.L. Rapid, sensitive, and selective copper (II) determination using sensitive chromogenic azo dye based on sulfonamide. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 247, 119103. [Google Scholar] [CrossRef]
- Popova, A.; Stamboliyska, B.; Velcheva, E. Experimental and DFT studies on the IR spectral and structural changes arising from the conversion of 4-amino-N-[amino(imino)methyl] benzenesulfonamide (sulfaguanidine) into azanion. Bulg. Chem. Commun. 2014, 46, 149–156. [Google Scholar]
- Akocak, S.; Taslimi, P.; Lolak, N.; Işık, M.; Durgun, M.; Budak, Y.; Türkeş, C.; Gülçin, İ.; Beydemir, Ş. Synthesis, Characterization, and Inhibition Study of Novel Substituted Phenylureido Sulfaguanidine Derivatives as α-Glycosidase and Cholinesterase Inhibitors. Chem. Biodivers. 2021, 18, e2000958. [Google Scholar] [CrossRef] [PubMed]
- Bazyl, O.K.; Chaikovskaya, O.N.; Chaidonova, V.S.; Bocharnikova, E.N.; Maier, G.V. Spectral-luminescent properties and the nature of electronically excited states of sulfaguanidine in water. Opt. Spectrosc. 2022, 130, 627–636. [Google Scholar] [CrossRef]
- Eagle, H.; Doak, G.O. The biological activity of arsenosobenzenes in relation to their structure. Pharmacol. Rev. 1951, 3, 107–143. [Google Scholar] [PubMed]
- Northey, E.H. The sulfonamides and allied compounds. J. Am. Pharm. Assoc. 1948, 37, 338–352. [Google Scholar] [CrossRef]
- Rinco, O.; Kleinman, M.H.; Bohne, C. Reactivity of Benzophones in the Different Binding Sites of Sodium Cholate Aggregates. Langmuir 2001, 17, 5781–5790. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Morev, A.V. Investigation of the spectral-luminescent and physico-chemical properties of monosubstituted aniline molecules. Actual Probl. Mod. Sci. 2007, 36, 112–113. [Google Scholar]
- Artyukhov, V.Y.; Morev, A.V.; Morozova, Y.P.; Pomogaev, V.A. Investigation of the Heavy-Atom Effect on the Spectral-Luminescent Properties of Dichloroanilines. Russ. Phys. J. 2002, 45, 1203–1207. [Google Scholar] [CrossRef]
- Sverdlova, O.V. Elektronnye Spektry v Organicheskoi Khimii (Electron Spectra in Organic Chemistry); Khimiya: Leningrad, Russia, 1973; 248p. [Google Scholar]
- Tchaikovskaya, O.N.; Chaidonova, V.S.; Ashmarina, M.V. Absorption and fluorescence spectra of sulgin in the presence of methylene blue. Izv. Vuzov. Phys. 2021, 64, 123–128. [Google Scholar] [CrossRef]
- Herzberg, G. Electronic Structure and Structure of Polyatomic Compounds; Mir: Moscow, Russia, 1969. [Google Scholar]
- Brand, J.C.D.; Williams, D.R.; Cook, T.J. Vibrational analysis of the first ultraviolet band system of aniline. J. Mol. Spectrosc. 1966, 20, 359–380. [Google Scholar] [CrossRef]
- Khudozhitkov, A.E.; Stange, P.; Golub, B.; Paschek, D.; Stepanov, A.G.; Kolokolov, D.I.; Ludwig, R. Characterization of doubly ionic hydrogen bonds in protic ionic liquids by NMR deuteron quadrupole coupling constants: Differences to H-bonds in amides, peptides, and proteins. Angew. Chem. Int. Ed. 2017, 56, 14310–14314. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, V.G.; Alfimov, M.V. Molecular Photonics. In Fundamental and Applied Research; Results and Prospects 1996–2013; Center for Photochemistry RAS: Moscow, Russia, 2013. [Google Scholar]
- Artyukhov, V.Y.; Galeeva, A.I. Spectroscopic parametrization of the CPDP method. Izv. Univ. Phys. 1986, 29, 96–100. [Google Scholar]
- Bocharnikova, E.N.; Tchaikovskaya, O.N.; Bazyl, O.K.; Artyukhov, V.Y.; Mayer, G.V. Chapter Seven—Theoretical study of bisphenol A photolysis. Adv. Quantum Chem. 2020, 81, 191–217. [Google Scholar] [CrossRef]
- Bonassorsi, R.; Pullman, A.; Scroco, T.; Nomasi, J. The molecular electrostatic potentials for the nucleic acid bases: Adenine, thymine, and cytosine. Theoret. Chem. Acta 1972, 24, 51–60. [Google Scholar] [CrossRef]
- Scroco, T.; Nomasi, J. Electronic Molecular Structure, Reactivity and Intermolecular Forces: An Heuristic Interpretation by Means of Electrostatic Molecular Potentials. Adv. Quantum Chem. 1978, 11, 115–193. [Google Scholar] [CrossRef]
- Komarov, P.V.; Plotnikov, V.G. Intermolecular Interactions and Spectral-Luminescent Properties of Polyatomic Molecules. In Fundamental and Applied Research; Results and Prospects 1996–2013; Center for Photochemistry RAS: Moscow, Russia, 2013. [Google Scholar]
- Plotnikov, V.G.; Smirnov, V.A.; Alfimov, M.V. Two-photon photoprocesses in molecular systems. High Energy Chem. 2009, 43, 241–256. [Google Scholar] [CrossRef]
- Mayer, G.V.; Artyukhov, V.Y.; Karypov, A.V. Spin orbital interaction of aromatic-molecule ππ-states. Opt. Spectrosc. 1989, 66, 823–826. [Google Scholar]
- Mayer, G.V.; Plotnikov, V.G.; Artyukhov, V.Y. Nature of electronically excited states of organic compounds and processes of nonradiative conversion. Russ. Phys. J. 2016, 59, 513–524. [Google Scholar] [CrossRef]
- Bridges, J.W.; Williams, R.T. Fluorescence of Some Substituted Benzenes. Nature 1962, 4849, 59–61. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | Structure of Aniline | |||||
|---|---|---|---|---|---|---|
| Flat Structure | Nitrogen Atom Out of Plane, ZN = 0.25 Å | |||||
| Type | ν, cm−1 (λ, nm) | f | Type | ν, cm−1 (λ, nm) | f | |
| Isolated molecule | ||||||
| S1 | ππ * | 35,460 (282) | 0.06 | ππ * | 36,460 (274) | 0.01 |
| S2 | ππ * | 39,550 (253) | 0.06 | ππ * | 39,840 (250) | 0.13 |
| S4 | - | - | - | ππ * | 46,800 (214) | 0.48 |
| S6 | ππ * | 48,230 (207) | 0.62 | πσ * | 48,990 (204) | 0.14 |
| S7 | ππ * | 48,470 (206) | 0.74 | ππ * | 49,110 (203) | 0.72 |
| Aniline + H2O | ||||||
| S1 | ππ * | 35,890 (279) | 0.04 | ππ * + πσ * | 36,730 (272) | 0.01 |
| S2 | ππ * | 40,410 (248) | 0.17 | ππ * | 40,780 (245) | 0.14 |
| S4 | - | - | - | πσ * | 47,200 (211) | 0.44 |
| S6 | ππ * | 48,410 (206) | 0.62 | ππ * | 49,000 (204) | 0.79 |
| S7 | ππ * | 48,470 (206) | 0.76 | ππ * + πσ * | 49,330 (203) | 0.17 |
| Aniline + H+H2O | ||||||
| S1 | ππ * | 36,960 (270) | 0.02 | ππ * | 37,100 (270) | 0.00 |
| S2 | ππ * | 42,410 (236) | 0.14 | ππ * | 41,960 (238) | 0.11 |
| S4 | - | - | - | ππ * + πσ * | 46,940 (213) | 0.31 |
| S5 | ππ * | 48,880 (205) | 0.74 | ππ * | 49,380 (202) | 0.82 |
| S6 | ππ * | 49,120 (204) | 0.79 | ππ * + πσ * | 49,740 (201) | 0.39 |
| Fragment | Flat Aniline | Aniline with Nitrogen Atom Out of Plane, ZN = 0.25 Å | ||||
|---|---|---|---|---|---|---|
| S0 | S1 | S2 | S0 | S1 | S2 | |
| Isolated molecule | ||||||
| –NH2 | −0.045 | 0.143 | 0.135 | −0.084 | 0.004 | 0.029 |
| –C6H5 | 0.045 | −0.143 | −0.135 | 0.084 | −0.004 | −0.029 |
| Aniline + H2O | ||||||
| –NH2 | −0.061 | 0.128 | 0.131 | −0.082 | 0.009 | 0.116 |
| –C6H5 | 0.091 | −0.102 | −0.105 | 0.115 | −0.020 | −0.012 |
| H2O | −0.031 | −0.026 | −0.031 | −0.033 | −0.028 | −0.026 |
| Aniline + H+H2O | ||||||
| S0 | S1 | S3 | S0 | S1 | S3 | |
| –NH2 | −0.104 | 0.034 | −0.275 | −0.110 | −0.094 | −0.079 |
| –C6H5 | 0.120 | 0.137 | 0.288 | 0.175 | 0.220 | 0.148 |
| H+H2O | 0.985 | 0.828 | 0.983 | 0.934 | 0.874 | 0.931 |
| Calculation | Experiment | ||||
|---|---|---|---|---|---|
| State | Ei, cm−1 | λ, nm | f | νmax, cm−1 | λmax, nm |
| Sulfaguanidine + 2H2O | |||||
| S1 (ππ *) | 35,240 | 284 | 0.05 | 34,500 | 290 |
| S2 (πσ *) | 38,450 | 260 | 0.00 | 38,600 | 259 |
| S3 (ππ *) | 39,110 | 256 | 0.40 | ||
| S9 (ππ * + σσ *) | 47,330 | 211 | 1.00 | 48,100 | 208 |
| S10 (ππ *) | 47,830 | 209 | 0.44 | ||
| S11 (σσ *) | 48,269 | 207 | 0.10 | ||
| Sulfaguanidine + 2H+H2O | |||||
| S1 (ππ *) | 34,680 | 288 | 0.06 | 34,500 | 290 |
| S2 (πσ *) | 35,300 | 283 | 0.01 | ||
| S3 (ππ *) | 38,300 | 261 | 0.47 | 39,100 | 256 |
| S8 (ππ *) | 47,230 | 212 | 0.51 | 48,350 | 207 |
| S9 (ππ * + σσ *) | 47,780 | 209 | 0.97 | ||
| S10 (ππ * + σσ *) | 50,060 | 200 | 0.12 | 50,000 | 200 |
| S17 (σσ *) | 53,850 | 186 | 0.14 | ||
| Fragment, Group | Electronic State | ||||
|---|---|---|---|---|---|
| S0 | S1 | S3 | S9 | S10 | |
| Sulfaguanidine | |||||
| Aniline: | 0.081 | 0.048 | 0.075 | 0.019 | 0.048 |
| NH2 | −0.044 | 0.134 | 0.130 | −0.027 | −0.014 |
| C6H4 | 0.125 | −0.086 | −0.055 | 0.046 | 0.062 |
| Sulfonic | 0.001 | 0.022 | 0.019 | 0.087 | 0.040 |
| Guanidine: | −0.080 | −0.070 | −0.089 | −0.103 | −0.088 |
| N11=C12 | −0.074 | −0.065 | −0.085 | −0.124 | −0.085 |
| 2NH2 | −0.006 | −0.005 | −0.004 | 0.021 | −0.003 |
| Sulfaguanidine + 2H2O | |||||
| Aniline: | 0.092 | 0.068 | 0.076 | −0.118 | 0.050 |
| NH2 | −0.040 | 0.134 | 0.149 | −0.036 | −0.004 |
| C6H4 | 0.132 | −0.066 | −0.073 | 0.154 | 0.054 |
| Sulfonic | 0.063 | 0.079 | 0.083 | 0.153 | 0.142 |
| Guanidine: | −0.070 | −0.061 | −0.075 | 0.046 | −0.110 |
| N11=C12 | −0.078 | −0.071 | −0.086 | −0.093 | −0.121 |
| 2NH2 | 0.008 | 0.009 | 0.013 | 0.139 | 0.011 |
| 2H2O | −0.087 | −0.086 | −0.085 | −0.084 | −0.083 |
| Sulfaguanidine + 2H+H2O | |||||
| S0 | S1 | S3 | S8 | S9 | |
| Aniline: | 0.170 | 0.205 | 0.339 | 0.349 | 0.096 |
| NH2 | −0.001 | 0.240 | 0.256 | 0.050 | 0.005 |
| C6H4 | 0.171 | −0.035 | 0.083 | 0.299 | 0.091 |
| Sulfonic | −0.123 | −0.121 | −0.142 | −0.136 | −0.012 |
| Guanidine: | −0.030 | −0.066 | −0.178 | −0.166 | 0.064 |
| N11=C12 | −0.043 | −0.073 | − | −0.159 | −0.112 |
| 2NH2 | 0.013 | 0.007 | −0.005 | −0.007 | 0.048 |
| 2H + H2O | 1.983 | 1.983 | 1.983 | 1.983 | 1.981 |
| RSi | Atom Number in Bonds | |||||||
|---|---|---|---|---|---|---|---|---|
| 1–2 | 2–3 | 3–4 | 4–5 | 5–6 | 6–1 | 1–7 | 4–8 | |
| RS0 | 1.40 | 1.40 | 1.40 | 1.40 | 1.40 | 1.40 | 1.409 | 1.783 |
| Isolated molecule | ||||||||
| RS3 | 1.466 | 1.390 | 1.489 | 1.493 | 1.393 | 1.470 | 1.402 | 1.806 |
| Sulfaguanidine + 3H2O | ||||||||
| RS3 | 1.463 | 1.388 | 1.480 | 1.487 | 1.382 | 1.470 | 1.397 | 1.794 |
| Sulfaguanidine + 2H+H2O | ||||||||
| RS3 | 1.460 | 1.390 | 1.464 | 1.497 | 1.397 | 1.465 | 1.397 | 1.803 |
| Absorbance | Fluorescence | |||||||
|---|---|---|---|---|---|---|---|---|
| The Quasi-Quinoid Structure with the Nitrogen Atom in the Phenyl Plane | The Quasi-Quinoid Structure with the Nitrogen Atom Outside the Phenyl Plane | |||||||
| State | Ei, cm−1 | f | State | Ei, cm−1 | f | State | Ei, cm−1 | f |
| S1 (ππ *) | 35,250 | 0.045 | S1 (πσ *) | 28,910 | 0.005 | S1 (ππ *) | 28,920 | 0.389 |
| S2 (πσ *) | 38,390 | 0.035 | S2 (ππ *) | 30,850 | 0.578 | S2 (πσ *) | 32,530 | 0.035 |
| S3 (ππ *) | 38,770 | 0.307 | S3 (ππ *) | 33,120 | 0.064 | S3 (ππ *) | 33,080 | 0.006 |
| Calculation | Experiment * | |||||||
|---|---|---|---|---|---|---|---|---|
| Efl, cm−1 (nm) | ΔνST, cm−1 | kr, s−1 | kIC, s−1 | kST, s−1 | φ | Efl, cm−1 (nm) | ΔνST, cm−1 | φ |
| Isolated molecule | 28,570 (350) | 9800 | 0.16 | |||||
| 28,920 (352) | 10,350 | 2 × 108 | 7 × 104 | 1 × 1010 | 0.02 | |||
| Sulfaguanidine + 3H2O | ||||||||
| 28,210 (354) | 10,360 | 1 × 108 | 2 × 105 | 6 × 108 | 0.18 | |||
| Sulfaguanidine + H2O + 2(H+H2O) q = +2e | ||||||||
| 28,360 (353) | 9510 | 2 × 108 | 1 × 105 | 5 × 109 | 0.03 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tchaikovskaya, O.; Bocharnikova, E.; Bazyl, O.; Chaidonova, V.; Mayer, G.; Avramov, P. Nature of Luminescence and Pharmacological Activity of Sulfaguanidine. Molecules 2023, 28, 4159. https://doi.org/10.3390/molecules28104159
Tchaikovskaya O, Bocharnikova E, Bazyl O, Chaidonova V, Mayer G, Avramov P. Nature of Luminescence and Pharmacological Activity of Sulfaguanidine. Molecules. 2023; 28(10):4159. https://doi.org/10.3390/molecules28104159
Chicago/Turabian StyleTchaikovskaya, Olga, Elena Bocharnikova, Olga Bazyl, Vlada Chaidonova, George Mayer, and Paul Avramov. 2023. "Nature of Luminescence and Pharmacological Activity of Sulfaguanidine" Molecules 28, no. 10: 4159. https://doi.org/10.3390/molecules28104159