4.2. Methods
Chromatography. Ultramate 3000 HPLC system (DIONEX, Sunnyvale, USA). equipped with a photodiode array detector (with fixed wavelengths of 210, 270, 310, and 330 nm) was used to determine the purity of products. The method consisted of a gradient elution from 20% to 90% aqueous acetonitrile using a Hypersil Gold (5μm particle size) reverse-phase column at 25 °C. HPLC data were acquired and processed using Chromeleon 6.8 software (DIONEX, Sunnyvale, CA, USA). Chromatography for purification of products was performed on a 230–400 mesh silica-gel Sigma Aldrich (Steinheim, Germany) filled glass column. Analytical TLC was performed on F254 silica gel plates purchased from Sigma Aldrich. Compounds were visualized using UV light (254 nm) and/or via staining with 0.05% w/v palladium chloride solution in MeOH/HCl.
NMR spectroscopy. 1H, 11B, 11B{1H}, 13C{1H}, 31P, and 31P{1H} NMR spectra were recorded with a Bruker Advance III 600 MHz spectrometer.
UV-Vis spectrophotometry and line-fitting analysis. Measurements were performed using a Jasco V-750 UV spectrophotometer at room temperature in acetonitrile.
MS and FT-IR. MALDI-TOF MS spectra were recorded using a Voyager–Elite mass spectrometer (PerSeptive Biosystems) with 3-hydroxypicolinic acid (HPA) as the matrix. ESI MS mass spectra were recorded using Agilent 6546 LC/Q-TOF (Santa Clara, Kalifornia, United States). Negative ions were detected. Infrared absorption spectra were recorded using a Nicolet 6700 FT-IR spectrometer (Thermo Scientific) equipped with a Smart orbit diamond Attenuated Total Reflectance (ATR) accessory. Samples to be analyzed were placed on a diamond ATR element in a solid form or through casting from CH2Cl2 solution. Data were acquired and processed using Omnic 8.1 software (Thermo Scientific, Waltham, CA, USA).
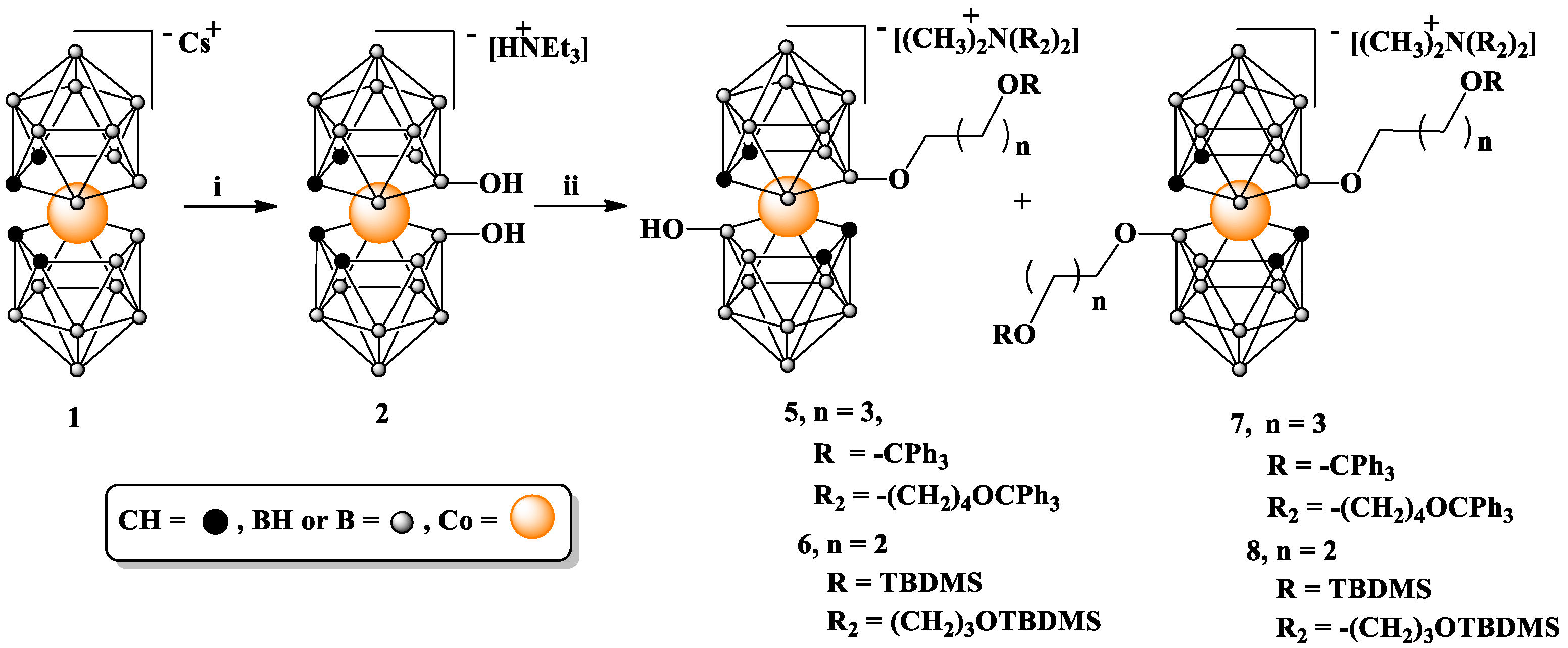
8,8′-dihydroxy-bis(1,2-dicarbollido)-3-cobalt(1-)ate HNEt3 (
2) was synthesized according to the procedure reported by Plešek et al. [
22].
4-Trityloxybutyl 4-methylbenzenesulfonate (
3) was obtained as described in [
39].
3-Bromo-1-trityloxypropane (
14) was synthesized as described previously [
10].
1,3-bis(trityloxy)propan-2-ol (
18) was synthesized according to a procedure in the literature [
31].
1,4-bis-(4-methylbenzenesulfonate)butane (
19) was synthesized according to a modified procedure from the literature [
40].
Synthesis of 3,3′-Co{[(8-O(CH2)4OCPh3]-1,2-C2B9H10}(8′-OH-1,2-C2B9H10) (CH3)2N[(CH2)4OPh3]2 (5) and 3,3′-Co[(8-O(CH2)4OCPh3-1,2-C2B9H10)]2 (CH3)2N[(CH2)4Oph3]2 (7). Compound 2 (50 mg, 0.10 mmol) was dissolved in 0.5 mL of anhydrous dimethoxyethane (DME) and added to 60% NaH in mineral oil dispersion (4.4 mg, 0.10 mmol NaH) under argon atmosphere. The reaction mixture was stirred for 2 h at room temperature. After this time has elapsed, the solvent was evaporated under reduced pressure, and the resultant solid residue was dissolved in 0.5 mL of anhydrous dimethylformamide (DMF) and added to a second aliquot of 60% NaH in mineral oil dispersion (26.2 mg, 0.65 mmol NaH). After sitting for 2 h at room temperature, the mixture was dropped into a solution of 3 (318.8 mg, 0.65 mmol) in 0.5 mL of anhydrous DMF; then, the reaction mixture was heated at 80 °C (oil bath temperature) for 24 h. The post-reaction mixture was concentrated; then, 3 mL of H2O was added. The resulting precipitate was washed with several aliquots of H2O. Then, the solid was dissolved in 5 mL of CH2Cl2, and the solution was dried with anhydrous Na2SO4, filtered, and concentrated. Repeated column chromatography on silica gel using a gradient of CH3OH in CH3Cl (0 to 20%) as an eluting solvent system yielded 5 and 7 as the first and second bands, respectively. Fractions containing compound 7 were collected, evaporated to dryness, and then crystallized from hexane, furnishing 96% pure product as determined by HPLC. Monosubstituted compound 5 was obtained as a red oil.
(5): Yield: 7 mg (5%); TLC (CHCl3:MeOH 4:1); Rf: 0.53; MALDI-MS (m/z): 670.5 (calc. for C27B18H44O3Co1: 670.17). Since excessively small quantities of this product were obtained, it was not further analyzed via NMR.
(7): Yield: 180 mg (62%); TLC (CHCl3:MeOH 4:1); Rf: 0.64; 1H NMR (500 MHz, CD3CN) δ: 7.43 (m, 24H, Harom), 7.32 (m, 24H, Harom), 7.25 (m, 12H, Harom), 4.18 (s, 4H, CHcarborane), 3.31 (t, J = 6.2 Hz, 4H, BOCH2CH2CH2CH2OTr), 3.07 (m, 8H, CH2OTr), 2.99 (t, J = 6.4 Hz, 4H, NCH2CH2CH2CH2OTr), 2.86 (s, 6H, N(CH3)2), 1.68 (m, 4H, NCH2CH2CH2CH2OTr), 1.60 (m, 8H, CH2CH2OTr), and 1.48 (m, 4H, BOCH2CH2CH2CH2OTr); 13C{1H} NMR (126 MHz, CD3CN) δ: 145.53 and 145.18 (12C, aromatictrityl); 129.44 and 129.41 (24C, aromatictrityl); 129.37 and 128.83 (24C, aromatictrityl); 128.21 (12C, aromatictrityl); 87.43 and 87.06 (4C, C(Ph)3), 69.43 (2C, BOCH2CH2CH2CH2OTr), 64.30 (2C, BOCH2CH2CH2CH2OTr), 63.30 (2C, NCH2CH2CH2CH2OTr), 62.33 (2C, NCH2CH2CH2CH2OTr), 51.56 (4C, CHcarb), 30.39, 29.68 (4C, CH2CH2OTr), 27.51, 27.13 (2C, BOCH2CH2CH2CH2OTr), and 20.29 (NCH2CH2CH2CH2OTr); 11B{1H} NMR (160 MHz, CD3CN) δ: 20.63 (s, 2B, B8,8′), −3.56 (s, 2B, B10,10′), −7.52 (s, 4B, B4,4′,7,7′), −9.03 (s, 4B, B9,9′,12,12′), −20.53 (s 4B, B5,5′,11,11′), and −28.38 (s, 2B, B6,6′) 11B NMR (160 MHz, CD3CN) δ: 20.66 (s, 2B, B8,8′), −3.59 (d, 2B, B10,10′), −8.22 (m, 8B, B9,9′,12,12′,4,4′,7,7′), −20.48 (d, 2B, B5,5′,11,11′), and −28.09 (d, 2B, B6,6′); FT-IR (cm−1): 3083.53, 3055.16, 3029.62 (ν C-H aromatic, (C-Hcarb); 2927.18 (ν C-Hasym, CH2); 2867.43 (ν C-H, CH2O, ν C-Hsym, CH2); 2543.23 (ν B-H); 1596.06, 1488.72, 1447.49 (ν C=C); 1152.23; 745.10 and 693.37 (δ C-H aromatic); 703.89 (ν B-B); MALDI-MS (m/z): found 984.2 (calc. for C50B18H66O4Co1: 984.59).
Synthesis of 3,3′-Co [8-O(CH2)3OTBDMS]-1,2-C2B9H10)(8′-OH-1,2-C2B9H10) (CH3)2N[(CH2)3OTBDMS)]2 (6) and 3,3′-Co[8-O(CH2)3OTBDMS-1,2-C2B9H10)]2 (CH3)2N[(CH2)3OTBDMS)]2 (8). Compound
2 (300 mg, 0.65 mmol) was dissolved in 3 mL of anhydrous DME under argon atmosphere and added to 60% NaH in mineral oil dispersion (26.2 mg, 0.65 mmol NaH). The reaction mixture was stirred for 2 h at room temperature. Subsequently, the solvent was evaporated, and the solid residue was dissolved in 3 mL of anhydrous DMF and added to a second portion of 60% NaH in mineral oil dispersion (157 mg, 3.93 mmol NaH). After sitting for 2 h at room temperature, the mixture was heated to 60 °C (oil bath temperature) and 911 μL of (3-bromopropoxy)(
tert-butyl)dimethylsilane (
4) (3.93 mmol) was added dropwise. After 22 h, an additional quantity of
4 (600 μL, 2.59 mmol) was added, and the mixture was stirred for next 24 h at 60 °C. A white solid was formed. The post-reaction mixture was then filtered through syringe filter (5 μm, PTFE, Carl Roth), and DMF was evaporated under reduced pressure. The solid residue was treated with 2 mL of CHCl
3 and filtered; then, the the filtrate was concentrated and loaded on a silica gel column for separation of products. First, 100% CHCl
3 and then 5% and 10% CH
3CN in CHCl
3 were used as eluting solvent systems. Compound
8 was isolated as first fraction and obtained in the form of orange crystals after solvent evaporation; its purity was above 95% as determined via HPLC. The second fraction containing
6 was obtained as red oil after solvent evaporation. Both products
6 and
8 were isolated as N,N-bis[(3-(
tert-butyldimethylsilyloxypropyl)]-N,N-dimethyl ammonium salts [
26].
(6): Yield: 60 mg (10%); TLC (CHCl3:CH3CN 3:1) Rf: 0.53; 1H NMR (600 MHz, CD3CN) δ: 6.05 (s, 1H, OH), 3.73 (t, J = 6.5 Hz, 2H, BOCH2CH2CH2OSi), 3.69 (m, 4H, NCH2CH2CH2OSi), 3.65 (m, 2H, BOCH2CH2CH2OSi), 3.56 (s, 2H, CHcarb), 3.45 (s, 2H, CHcarb), 3.27 (m, 4H, NCH2CH2CH2OSi), 2.97 (s, 6H, N(CH3)2), 1.88 (m, 4H, NCH2CH2CH2OSi), 1.74 (m, J = 6.4 Hz, 2H, BOCH2CH2CH2OSi), 0.90 (s, 9H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), 0.89 (s, 18H, NCH2CH2CH2OSi(CH3)2C(CH3)3), 0.07 (s, 6H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), and 0.05 (s, 12H, NCH2CH2CH2OSi(CH3)2C(CH3)3); 13C{1H} NMR (126 MHz, CD3CN) δ: 67.05 (1C, BOCH2CH2CH2OSi), 62.56 (2C, NCH2CH2CH2OSi), 61.23 (1C, BOCH2CH2CH2OSi), 60.17 (2C, NCH2CH2CH2OSi), 52.03 (2C, N(CH3)2), 46.23 (2C, CHcarb), 45.51 (2C, CHcarb), 35.92 (1C, BOCH2CH2CH2OSi), 26.45 (2C, NCH2CH2CH2OSi), 26.27 (6C, OSi(CH3)2C(CH3)3), 26.11 (3C, OSi(CH3)2C(CH3)3), 18.81 (1C, OSi(CH3)2C(CH3)3), 18.72 (2C, OSi(CH3)2C(CH3)3), −5.10 (2C, OSi(CH3)2C(CH3)3), and −5.40 (4C, OSi(CH3)2C(CH3)3); 11B{1H} NMR (193 MHz, CD3CN) δ: 27.16 (s, 1B, B8′), 25.10 (s, 1B, B8), −5.02 to −9.09 (10B, overlapped B9,9′,10,10′12,12′,4,4′,7,7′), −20.10 to −20.69 (4B, B5,5′,11,11′), and −29.18 to −30.16 (2B, B6,6′); 11B NMR (160 MHz, CD3CN) δ: 27.16 (s, 1B, B8′), 25.09 (s, 1B, B8), −3.22 to −9.40 (10B, overlapped B9,9′,10,10′12,12′,4,4′,7,7) −19.72 to −21.11 (4B, B5,5′,11,11′), and −28.81 to −30.49 (2B, B6,6′); FT-IR (cm−1): 3278.64, 2952.00, 2927.58, 2855.64, 2539.99, 1470.65, 1387.53, 1360.69, 1252.66, 1156.76, 1093.25, 1005.54, 972.16, 938.48, 880.24, 832.61, 774.98, 718.29, and 661.26; ESI-MS (m/z): found 530.39 and 472.35 [M-tButyl]− (calc. for C13B18H43Si1O3Co1: 529.09).
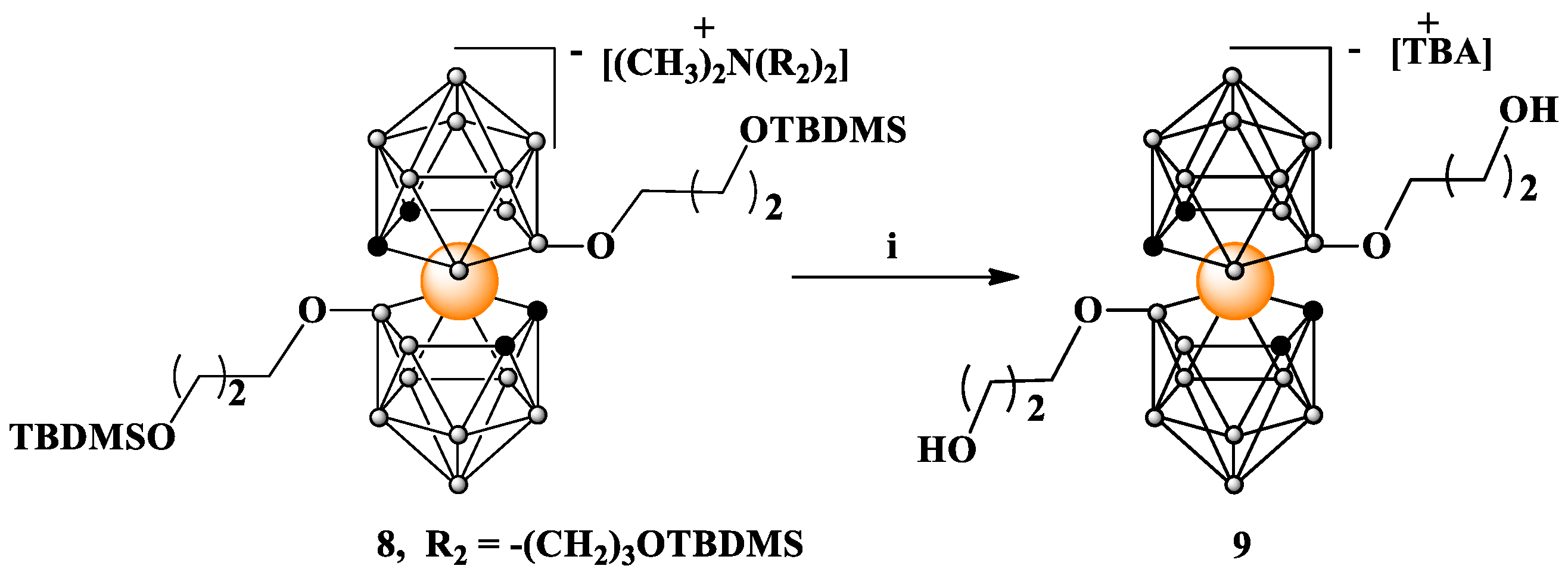
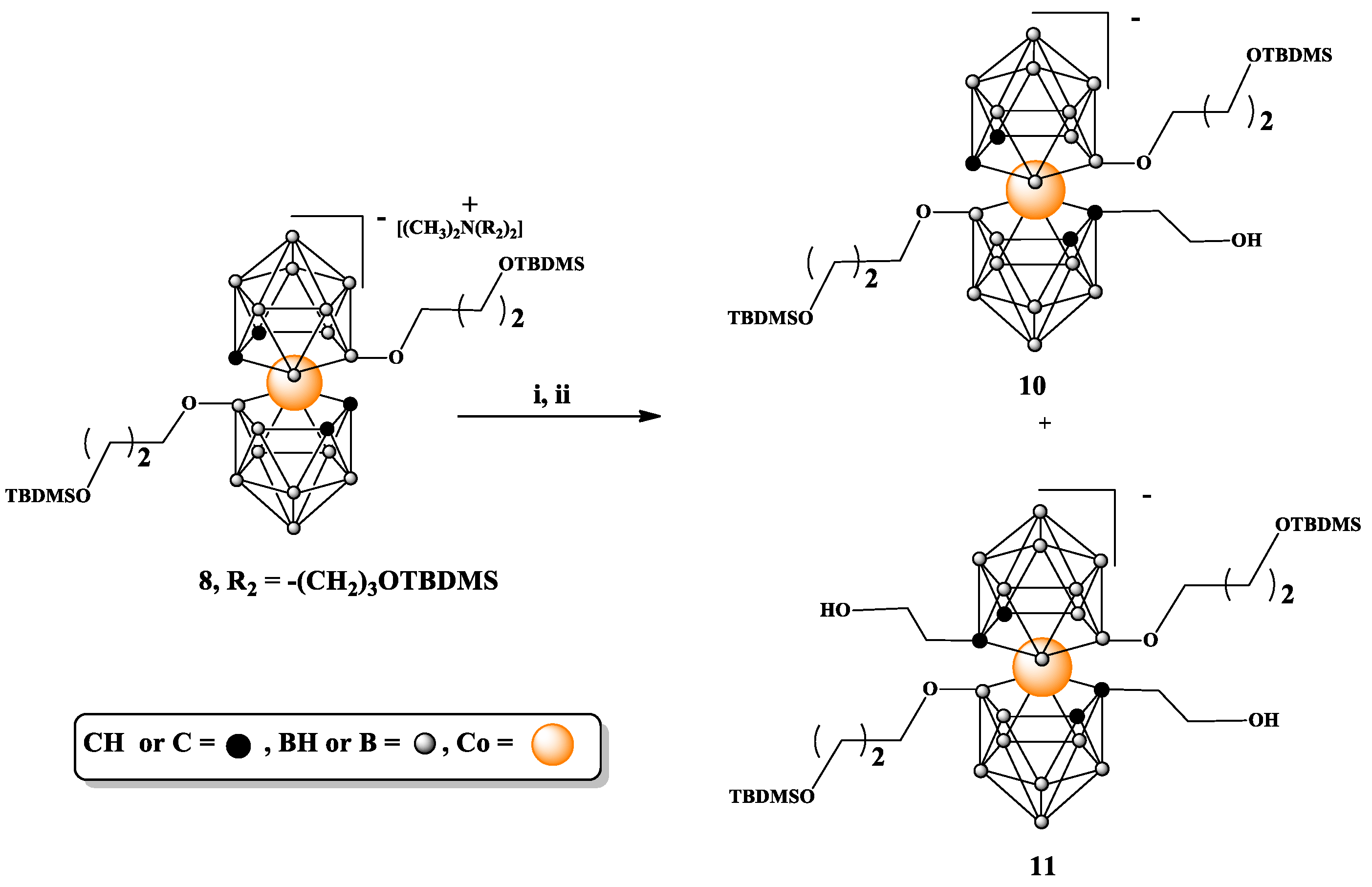
(8): Yield: 572 mg (80%); TLC (CHCl3:CH3CN 3:1) Rf 0.70; 1H NMR (500 MHz, CD3CN) δ: 4.18 (s, 4H, CHcarb), 3.69 (m, 4H, BOCH2CH2CH2OSi), 3.64 (t, J = 6.4 Hz, 4H, NCH2CH2CH2OSi), 3.38 (t, J = 6.0 Hz, 4H, and BOCH2CH2CH2OSi), 3.27 (m, 4H, NCH2CH2CH2OSi), 2.97 (s, 6H, N(CH3)2), 1.88 (m, 4H, NCH2CH2CH2OSi), 1.58 (m, J = 6.2 Hz, 4H, BOCH2CH2CH2OSi), 0.90 (s, 18H, OSi(CH3)2C(CH3)3), 0.88 (s, 18H, OSi(CH3)2C(CH3)3), 0.07 (s, 12H, OSi(CH3)2C(CH3)3), and 0.04 (s, 12H, OSi(CH3)2C(CH3)3); 13C NMR (126 MHz, CD3CN) δ: 66.18 (2C, BOCH2CH2CH2OSi), 62.54 (2C, NCH2CH2CH2OSi), 60.95 (2C, BOCH2CH2CH2OSi), 60.17 (2C, NCH2CH2CH2OSi), 52.06 (2C, N(CH3)2), 51.60 (4C, CHcarb), 35.97 (2C, BOCH2CH2CH2OSi), 26.46 (2C, NCH2CH2CH2OSi), 26.28 (6C, OSi(CH3)2C(CH3)3), 26.12 (6C, OSi(CH3)2C(CH3)3 18.83 (2C, OSi(CH3)2C(CH3)3), 18.72 (2C, OSi(CH3)2C(CH3)3), −5.09 (4C, OSi(CH3)2C(CH3)3), and −5.39 (4C, OSi(CH3)2C(CH3)3); 11B{1H} NMR (160 MHz, CD3CN) δ: 20.61 (s, 2B, B8,8′), −3.61 (s, 2B, B10,10′), −7.45 (s, 4B, B4,4′,7,7′), −9.03 (s, 4B, B9,9′,12,12′), −20.51 (s, 4B, B5,5′,11,11′), and −28.38 (s, 2B, B6,6′); 11B NMR (160 MHz, CD3CN) δ: 20.76 (s, 2B, B8,8′), −3.59 (d, 2B, B10,10′), −7.18 to −9.37 (m, 8B, overlapped B4,4′,7,7′,9,9′,12,12′), −20.48 (d, 4B, B5,5′,11,11′), and −28.02 (d, 2B, B6,6′); FT-IR (cm−1): 3048.6 (C-Hcarb); 2949.4 (ν C-Hasym, CH3); 2927.4 (ν C-Hasym, CH2), 2891.6 and 2884.2 (ν C-Hsym CH3); 2856.2 (ν C-H, CH2O, ν C-Hsym, CH2); 2605.2 and 2550.6 (ν B-H); 1470.8 (δ C-Hsym, CH2) and 1436.0; 1386.2; 1359.5; 1251.2 (Si-CH3) and 1161.9 (Si-O-C); 1093.1 (ν Si-O); 1019.3; 1006.3; 975.2; 955.3; 942.7; 881.; 831.8 and 772.4 (ν Si-C); 710.9; 661.0; MALDI-MS (m/z): found 700.5 (calc. for C22B18H62CoO4Si2 700.43).
Deprotection of compound 8 to obtain [3,3′-Co(8-O(CH2)3OH-1,2-C2B9H10)2] TBA (9). Compound 8 (20 mg, 0.018 mmol) was dissolved in 0.2 mL of tetrahydrofuran (THF). A total of 69 μL of terabutylammonium fluoride (TBAF) (1M solution in THF, 0.069 mmol) was added to the obtained solution, and the reaction mixture was stirred at room temperature overnight. Then, solvent was evaporated under reduced pressure, and the resulting oil was dissolved in CHCl3 and applied to the silica gel column prepared in the same solvent. Elution in a gradient 1 to 20% CH3CN in CHCl3 provided 96% pure 9 (determined by HPLC) as TBA salt. Yield: 12.5 mg (95%); TLC (CHCl3:CH3CN 3:1) Rf: 0.47; 1H NMR (500 MHz, CD3CN) δ: 4.17 (s, 4H, CHcarb), 3.52 (t, J = 6.3 Hz, 4H, BOCH2CH2CH2OH), 3.43 (t, J = 6.0 Hz, 4H, BOCH2CH2CH2OH), 3.10–3.03 (m, 8H, NCH2CH2CH2CH3), 2.58 (s, 2H, OH), 1.59 (m, J = 8.2, 3.7 Hz, 12H, BOCH2CH2CH2OH, NCH2CH2CH2CH3), 1.41–1.28 (m, 8H, NCH2CH2CH2CH3), and 0.96 (t, J = 7.4 Hz, 12H, NCH2CH2CH2CH3); 13C NMR (126 MHz, CD3CN) δ: 67.47 (2C, BOCH2CH2CH2OH), 60.63 (2C, BOCH2CH2CH2OH), 59.23 (4C, NCH2CH2CH2CH3), 51.39 (4C, CHcarborane), 35.54 (2C, BOCH2CH2CH2OH), 24.21 (4C, NCH2CH2CH2CH3), 20.25 (4C, NCH2CH2CH2CH3), and 13.72 (4C, NCH2CH2CH2CH3); 11B{1H} NMR (160 MHz, CD3CN) δ: 20.95 (s, 2B, B8,8′), −3.49 (s, 2B, B10,10′), −7.71 (s, 4B B4,4′,7,7′), −8.85 (s, 4B, B9,9′,12,12′), −20.44 (s, 4B, B5,5′,11,11′), and −28.14 (s, 2B, B6,6′); 11B NMR (160 MHz, CD3CN) δ: 20.97 (s, 2B, B8,8′), −3.48 (d, 2B, B10,10′), −7.18 to −9.26 (m, 8B, B4,4′7,7′9,9′,12,12′), −20.42 (d, 4B, B5,5′,11,11′), and −28.19 (d, 2B, B6,6′); FT-IR (cm−1): 3588.76 (ν O-H); 3450.79 (ν N+-R); 3052.83 (C-Hcarb); 2962.10 (ν C-Hasym, CH2); 3932.36 and 2873.70 (ν C-H, CH2O, ν C-Hsym, CH2); 2529.19 (ν B-H); 1467.75 (ν N+-R), 1380.63; 1161.25 (HO-C); 1105.54; 1062.05; 1007.15; 969.00; 945.10; 920.29; 876.90; 789.11; 735.69; 696.07; 664.83; MALDI-MS (m/z): found 472.8 (calc. for C10B18H34O4Co1 471.90).
Synthesis of 3,3′-Co{[8-O(CH2)3OTBDMS-1-(CH2)2OH]-1,2-C2B9H9)}[8′-O(CH2)3OTBDMS-1′,2′-C2B9H10)]− (10) and 3,3′-Co[(8-O(CH2)3OTBDMS-1-(CH2)2OH-1,2-C2B9H9)]2− (11). Compound 8 (50 mg, 0.04 mmol) was dried via co-evaporation with anhydrous benzene and then kept under vacuum, over P2O5 overnight. Then, it was dissolved in anhydrous DME (1 mL), and the solution was cooled in CO2/isopropanol cooling bath. After 15 min, n-BuLi (43 µL, 1.6 M solution in hexane, 1.5 eq) was added, and the reaction mixture was stirred for 10 min. Afterwards, cooling bath was removed, and the mixture was stirred for next 10 min. Then, the reaction mixture was cooled again in cooling bath and another portion of n-BuLi (43 µL) was added. After 15 min, ethylene oxide (60 µL, 2.9–3.1 M solution in THF, 4.5 eq) was added, and the reaction was left overnight in cooling bath. Then, CH2Cl2 (3 mL) was added to the reaction mixture, the reaction was quenched via addition of water, and the organic solution was washed three times with 5 mL portions of water. Organic layer was separated and dried over MgSO4; then, solvents were evaporated. Crude product was purified and mono- and bis-substituted products were separated through silica gel column chromatography using a gradient of MeOH in CH2Cl2 from 0 to 3% of MeOH.
(10): Yield: 4.7 mg (9%); TLC (MeOH:CH2Cl2 1:12.5): Rf: 0.28; ESI-MS (m/z): found 744.55, (calc. for C24B18H66O5Si2Co1 744.48). Since excessively small quantities of this product were obtained, it was not further analyzed via NMR.
(11): Yield: 15 mg (27%); TLC (MeOH:CH2Cl2 1:12.5): Rf: 0.16; 1H NMR (500 MHz, CD3CN) δ: 4.32–4.08 (s, 2H, diastereoizomeric CHcarborane), 3.77 to 3.52 (m, 16H, overlapped NCH2CH2CH2O, NCH2CH2CH2O, BOCH2CH2CH2O, BOCH2CH2CH2O), 3.52 to 3.39 (m, 4H, HOCH2CH2Ccarb), 3.39 to 3.31 (m, 4H, HOCH2CH2Ccarb), 3.03 (s, 6H, N(CH3)2), 1.74 (m, 4H, NCH2CH2CH2O), 1.65 (m, 4H, BOCH2CH2CH2O), 0.90 (s, 18H, NCH2CH2CH2OSi(CH3)2C(CH3)3), 0.88 (s, 18H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), 0.07 (s, 12H, BOCH2CH2CH2OSi(CH3)2C(CH3)3), and 0.05 (s, 12H, NCH2CH2CH2OSi(CH3)2C(CH3)3; 13C{1H} NMR (126 MHz, CD3CN) δ: 67.39 (2C, BOCH2CH2CH2O), 66.93 (1C, CHcarborane), 64.82 (1C, CHcarborane), 64.10 (2C, NCH2CH2CH2OSi), 62.28 (2C, BOCH2CH2CH2O), 61.00 (2C, HOCH2CH2Ccarb), 57.17 (2C, NCH2CH2CH2O), 56.24 (2C, N(CH3)2), 53.22 (2C, Ccarborane), 45.17 (2C, HOCH2CH2Ccarb), 36.89 (2C, NCH2CH2CH2O), 27.28 (2C, BOCH2CH2CH2O), 26.98 (6C, OSi(CH3)2C(CH3)3), 26.77 (6C, OSi(CH3)2C(CH3)3), 19.53 (2C, OSi(CH3)2C(CH3)3), 19.39 (2C, OSi(CH3)2C(CH3)3), −4.38 (4C, OSi(CH3)2C(CH3)3), and −4.73 (4C, OSi(CH3)2C(CH3)3). 11B{1H} NMR (120 MHz, CD3CN) δ: 29.65, 25.25, 24.28, 23.33, 21.98 (in ratio: 3:1.5:1:1:10), 31.27 to 19.66 (m, overlapped diastereoizomeric B8,8′), −2.63 to −13.75 (m, overlapped diastereoizomeric, B10,10′,9,9′,12,12′,4,4′,7,7′), −14.09 to −21.65 (m, overlapped diastereoizomeric B5,5′,11,11′), and −22.11 to −29.38 (m, overlapped diastereoizomeric B6,6′); 11B NMR (120 MHz, CD3CN) δ: 30.85–20.54 (m, overlapped diastereoizomeric B8,8′), −2.16 to −13.03 (m, overlapped diastereoizomeric, B10,10′,9,9′,12,12′,4,4′,7,7′), −13.91 to −21.45 (m, overlapped diastereoizomeric, B5,5′,11,11′), and −21.52 to −27.10 (m, overlapped diastereoizomeric, B6,6′); ESI-MS (m/z): found 788.58 (calc. for C26B18H70O6Si2Co1 788.53).
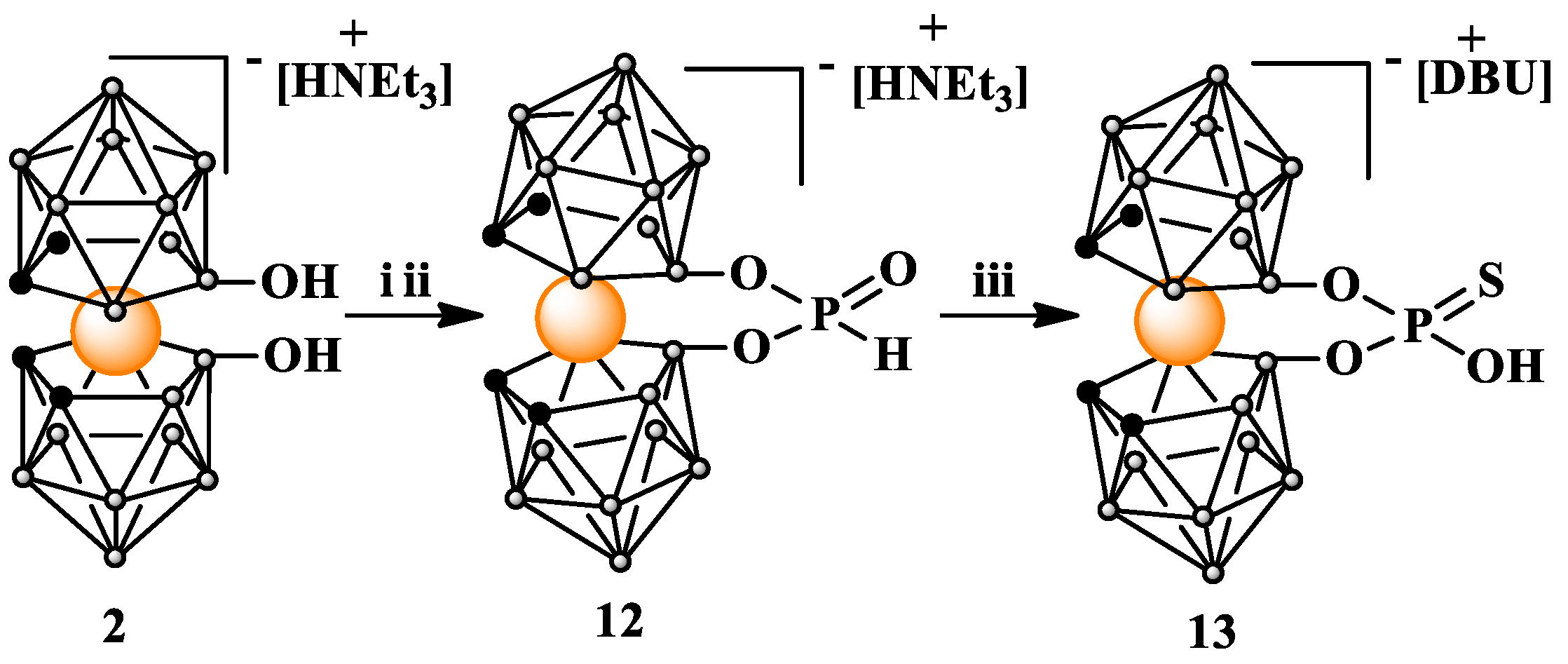
Synthesis of 8,8′-bridged [8,8′-O2P(O)H-3,3′-Co(1,2-C2B9H10)2] HNEt3 H-phosphonate (12). Imidazole (0.51 g, 7.5 mmol) was dissolved in minimum ammount of anhydrous acetonitrile. The solvent was evaporated under reduced pressure, and the procedure was repeated twice. After drying under vacuum for 1.5 h, imidazole was redissolved in 16 mL of anhydrous THF and the solution was cooled to −70 °C in a dry ice/isopropanol bath under argon atmosphere. PCl3 (210 µL, 2.40 mmol) was added dropwise followed by Et3N (1 mL, 7.17 mmol) mixed with 1 mL of anhydrous THF. The entire mixture was stirred at −70 °C for 15 min and then solution of 8,8′-dihydroxy-bis(1,2-dicarbollido)-3-cobalt(1-)ate HNEt3 (2) (320 mg, 0.69 mmol) in 13 mL of THF was added dropwise. After further 30 min, the reaction mixture was removed from cooling bath and allowed to warm to room temperature. After another 1 h, the reaction was quenched with 30 mL of water and extracted four times with 40 mL of diethyl ether (Et2O). The combined ether extracts were dried with MgSO4 and the solvent was evaporated. The resultant solid crude product was dryed under vacuum and then purified by silica gel (230–400 mesh) column chromatography using CH3CN:CHCl3 1:4 as eluting solvent system. Yield: 320 mg (91%); TLC (CH3CN:CHCl3 1:2); Rf: 0.35; 1H NMR (500 MHz, CD3CN) δ: 7.47, 6.07 (1H, P-H), 3.70 (s, 4H, CHcarborane), 3.10 (m, 6H, NCH2CH3), and 1.24 (t, 9H, NCH2CH3); 13C{1H} NMR (125 MHz, CD3CN) δ: 47.96 (4C, Ccarb), 47.74 (3C, NCH2CH3), and 9.24 (3C, NCH2CH3); 11B{1H} NMR (120 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8′), −2.83 (s, 2B, B10,10′), −5.70 (s, 4B, B9,9′,12,12′), −7.94 (s, 2B, B4,4′), −8.74 (s, 2B, B7,7′), −18.89 (s, 4B, B5,5′,11,11′), and −27.85 (s, 2B, B6,6′); 11B NMR (125 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8′), −2.83 (d, 2B, B10,10′), −5.70 (d, 4B, B9,9′,12,12′), −8.36 (t, 4B, B4,4′,7,7′), −18.91 (d, 4B, B5,5′,11,11′), and −27.84 (d, 2B, B6,6′); 31P{1H} NMR (202 MHz, CD3CN) δ: −3.01 (s, P-H); 31P NMR (202 MHz, CD3CN) δ: −3.00 (d, P-H); ATR-IR (cm−1): 3621, 3029, 2993, 2544, 1609, 1474, 1446, 1393, 1218, 1152, 1137, 1094, 1025, 993, 981, 920, 903, 871, 849, 787, 743, 691, and 666. UV-Vis λmax (nm): 297 and 445. ESI-MS (m/z): found 402.24 (calc. for C4H21O3B18P1Co1: 401.71).
Synthesis of 8,8′-bridged [8,8′-O2P(O)SH-3,3′-Co(1,2-C2B9H10)2] HDBU phosphorothioate (13). H-phosphonate acid ester 12 (130 mg, 0.26 mmol) was dissolved in anhydrous MeOH (6.5 mL). The solution was added under argon atmosphere to S8 (85 mg, 2.6 mmol). Then, 1,8-Diazabicyclo(5.4.0)undec-7-en (DBU) (160 µL, 1.05 mmol) was added, and the mixture was stirred for 96 h at room temperature. Subsequently, solvent was evaporated under reduced pressure. The crude product was dissolved in CH3CN and then purified via silica gel column chromatography using a gradient of CH3CN:CHCl3 from 1:4 to 1:1 as eluting solvent system. Finally, product 13 was eluted from the column using 100% MeOH as eluent. Yield: 105 mg (70%); TLC (CH3CN:CHCl3 2:1) Rf: 0.5; 1H NMR (500 MHz, CD3CN) δ: 9.14 (s, 1H, NH), 3.58 (s, 4H, CHcarborane), 3.50 (m, 2H, NHCH2CH2), 3.44 (t, J = 5.9 Hz, 2H, CH2NCH2CH2CH2NH), 3.31 (s, 2H, CH2NCH2CH2CH2NH), 2.69 (dd, J = 6.6, 3.5 Hz, 2H, NHCCH2), 1.97 (dd, J = 6.6 Hz, 2H, CH2NCH2CH2CH2NH), 1.72 (m, 4H, NHCCH2CH2CH2), and 1.65 (dt, J = 14.9, 5.2 Hz, 2H, NHCCH2CH2CH2CH2); 13C{1H} NMR (125 MHz, CD3CN) δ: 166.94 (NHCCH2), 54.98 (CH2NCH2CH2CH2NH), 49.26 (CH2NCH2CH2CH2NH), 46.76 (Ccarb), 46.63 (Ccarb), 39.04 (NHCH2), 33.42 (NHCCH2), 29.47 (NHCCH2CH2CH2), 27.05 (NHCCH2CH2CH2CH2), 24.52 (NHCCH2CH2CH2), and 19.96 (CH2NCH2CH2CH2NH); 11B{1H} NMR (120 MHz, CD3CN) δ: 23.46 (s, 2B, B8,8′), −3.54 (s, 2B, B10,10′), −5.72 (s, 4B, B9,9′,12,12′), −8.73 (s, 4B, B4,4′,7,7′), −19.49 (s, 4B, B5,5′,11,11′), and −28.25 (s, 2B, B6,6′); 11B NMR (120 MHz, CD3CN) δ: 23.46 (s, 2B, B8,8′), −3.50 (d, 2B, B10,10′), −5.74 (d, 4B, B9,9′,12,12′), −8.71 (d, 4B, B4,4′,7,7′), −19.53 (d, 4B, B5,5′,11,11′), and −28.34 (d, 2B, B6,6′); 31P{1H}NMR (202 MHz, CD3CN) δ: 48.63 (s); 31P NMR (202 MHz, CD3CN) δ: 48.62 (s); ATR-IR (cm−1): 3383 (ν OH), 3223 (ν NH), 3091 (ν NH), 3026, 2926 (ν CH), 2856 (ν CH), 2799 (ν CH), 2545 (ν BH), 1725, 1640, 1607, 1465, 1444, 1363, 1321, 1292, 1205, 1157, 1103, 1076, 978, 936, 910, 887, 836, 747, and 690. UV-Vis λmax (nm) 215, 296, and 450. ESI-MS (m/z): found: 434.20 (calc. for C4H21O3B18P1S1Co1 433.78).
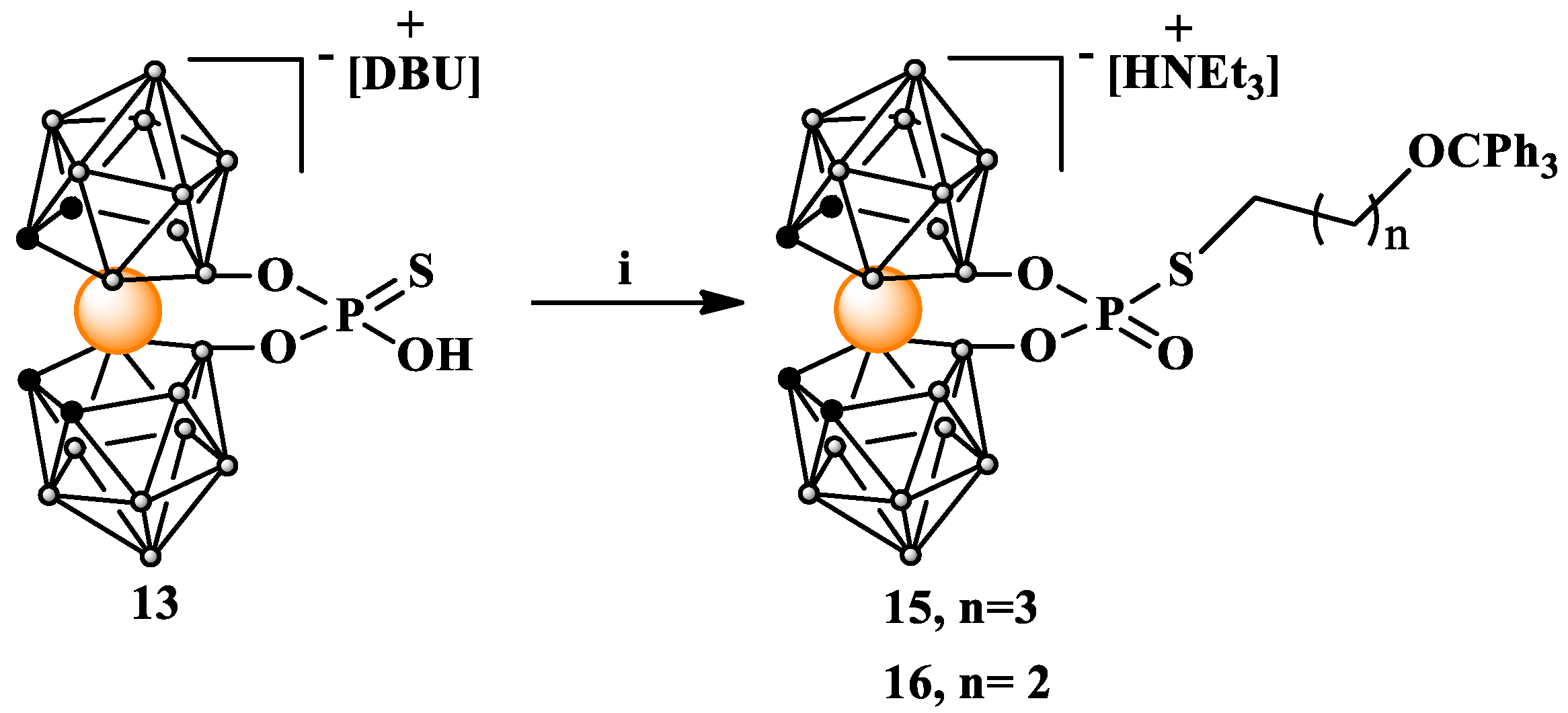
Synthesis of 8,8′-bridged [8,8′-O2P(O)S(CH2)nOCPh3-3,3′-Co(1,2-C2B9H10)2] HNEt3 S-alkylated phosphorothioates 15 and 16. [8,8′-O2P(O)SH-3,3′-Co(1,2-C2B9H10)2] HDBU (13) (13 mg, 0.02 mmol) was dissolved in acetone (0.520 mL); then, Et3N (65 µL, 0.46 mmol) was added under stirring at room temperature. The mixture was heated to 60 °C in an oil bath; then, alkylating agent 3 or 14 (0.04 mmol, dissolved in 130 µL of CH2Cl2) was added. The reaction mixture was maintained overnight at 60 °C with stirring; then, it was cooled to room temperature, and solvents were evaporated under reduced pressure. The residue was dispersed in CH2Cl2 and then filtered and the solution was loaded on silica gel column prepared in CH2Cl2. Chromatography was performed using a gradient of MeOH in CH2Cl2 from 0 to 3% MeOH. (15): Yield: 17 mg (90%); TLC (CH3CN:CHCl3 1:2): Rf: 0.71; 1H NMR (500 MHz, CD3CN): δ 7.41 (d, J = 7.5 Hz, 6H, Harom), 7.31 (t, J = 7.6 Hz, 6H, Harom), 7.23 (t, J = 7.2 Hz, 3H, Harom), 3.67 (s, 4H, CHcarborane), 3.07 (q, J = 7.3 Hz, 6H, NCH2CH3), 3.00 (t, J = 6.0 Hz, 2H SCH2CH2CH2CH2OTr), 2.78 (dt, 2H, SCH2CH2CH2CH2OTr), 1.72 (m, J = 7.1 Hz, 2H SCH2CH2CH2CH2OTr), 1.64 (m, J = 6.9 Hz, 2H, SCH2CH2CH2CH2Otr), and 1.21 (t, J = 7.3 Hz, 9H, NCH2CH3); 13C{1H} NMR (126 MHz, CD3CN): δ 145.55 (3C, aromatictrityl), 129.54 (6C, aromatictrityl), 128.87 (6C, aromatictrityl), 128.01 (3C, aromatictrityl), 87.30 (1C, OC(Ph)3), 63.90 (1C, SCH2CH2CH2CH2OTr), 47.76 (overlapped 3C, HNCH2CH3, 4C, CHcarborane), 31.20 (1C, SCH2CH2CH2CH2OTr), 29.75 (1C, SCH2CH2CH2CH2OTr), 28.84 (1C, SCH2CH2CH2CH2OTr), and 9.29 (1C, NCH2CH3); 11B{1H} NMR (160 MHz, CD3CN): δ 23.08 (s, 2B, B8,8′), −2.86 (s, 2B, B10,10′), −5.54 (s, 4B, B9,9′,12,12′), −8.36 (s, 4B, B4,4′,7,7′), −18.87 (s, 4B, B5,5′,11,11′), and −27.70 (s, 2B, B6,6′); 11B NMR (160 MHz, CD3CN): δ 23.06 (s, 2B, B8,8′), −2.87 (d, 2B, B10,10′), −5.57 (d, 4B, B9,9′,12,12′), −8.16 (d, 4B, B4,4′,7,7′), −18.92 (d, 4B, B5,5′,11,11′), and −27.63 (d, 2B, B6,6′); 31P NMR{1H} (202 MHz, CD3CN): δ 16.72 (s), 11.37 (s); 31P NMR (202 MHz, CD3CN): δ 16.72 (t) and 11.37 (t); ATR-IR (cm−1) 3032 (ν CH aromatic), 2987 (ν CH aliphatic), 2925 (ν CH aliphatic), 2851 (ν CH aliphatic), 2681, 2566 (ν BH), 1727, 1595, 1474, 1447, 1392, 1264, 1200, 1134, 1103, 1068, 1032, 1016, 980, 941, 917, 901, 849, 735 (aromatic CH bending), and 705 (aromatic CH bending). UV-Vis λmax (nm) 196, 299, and 440. ESI-MS (m/z): found: 748.37 (calc. for C27B18H43O4P1S1Co1: 748.24).
(16): Yield: 16 mg (86%); TLC (CH3CN:CHCl3 1:2) Rf: 0.69; 1H NMR (500 MHz, CD3CN) δ: 7.43 (m, 6H, Harom), 7.33 (m, 6H, Harom), 7.25 (m, 3H, Harom), 3.67 (s, 4H, CHcarborane), 3.10 (t, J = 6.0 Hz, 2H SCH2CH2CH2OTr), 3.08 (q, J = 7.3 Hz, 6H, NCH2CH3), 2.97 (dt, J = 14.9, 7.3 Hz, 2H, PSCH2CH2CH2OTr), 2.58 (s, 1H, CH3OH), 1.27 (t, J = 5.4 Hz, 2H PSCH2CH2CH2OTr), 1.22 (t, J = 7.3 Hz, 9H, NCH2CH3), and 1.18 (s, 3H, CH3OH); 13C{1H} NMR (126 MHz, CD3CN) δ: 145.27 (3C, aromatictrityl), 129.45 (6C, aromatictrityl), 128.78 (6C, aromatictrityl), 127.94 (3C, aromatictrityl), 87.25 (OC(Ph)3), 62.71 (PSCH2CH2CH2OTr), 55.13 (4C, CHcarb), 47.64 (HNCH2CH3), 47.59, 32.14, 31.98 (d, J = 6.1 Hz), 30.29, 29.66, 28.41 (d, J = 3.8 Hz), and 9.17 (HNCH2CH3); 11B{1H} NMR (160 MHz, CD3CN): δ (ppm) 23.08 (s, 2B, B8,8′), −2.86 (s, 2B, B10,10′), −5.53 (s, 4B, B9,9′,12,12′), −8.36 (s, 4B, B4,4′,7,7′), −18.87 (s, 4B, B5,5′,11,11′), and −27.70 (s, 2B, B6,6′); 11B NMR (160 MHz, CD3CN): δ 23.07 (s, 2B, B8,8′), −2.92 (d, 2B, B10,10′), −5.60 (d, 4B, B9,9′,12,12′), −8.38 (d, 4B, B4,4′,7,7′), −18.98 (d, 4B, B5,5′,7,7′), and −27.88 (d, 2B, B6,6′); 31P{1H} NMR (202 MHz, CD3CN) δ: 16.54 (s); 31P NMR (202 MHz, CD3CN) δ: 16.54 (t); ATR-IR (cm−1) 3032 (ν CH aromatic), 2987 (ν CH aliphatic), 2925 (ν CH aliphatic), 2851 (ν CH aliphatic), 2684, 2567 (ν BH), 1699, 1596, 1474, 1447, 1392, 1265, 1200, 1135, 1104, 1066, 1032, 1016, 980, 941, 917, 902, 849, 735 (aromatic CH bending), and 705 (aromatic CH bending). UV-Vis λmax (nm) 194, 299, and 436. ESI-MS (m/z): 734.35 (calc. for C26B18H41O4P1S1Co1 734.17).
Synthesis of 4-[1,3-bis(trityloxy)propan-2-yl-oxy]butyl-4-methylbenzenesulfonate (20). The reaction was performed under argon atmosphere in anhydrous conditions. 1,3-Bis(trityloxy)propan-2-ol (
18) (2.35 g, 4.07 mmol) was dissolved in 18 mL of anhydrous DMF; then, NaH
60% (195 mg, 4.87 mmol) was added. After stirring for 15 min, 1,4-bis(
p-toluenesulfonyloxy)butane [
40] (4.23 g, 10.63 mmol), dissolved in 18 mL of DMF, was added. The reaction mixture was stirred for another 2 h at room temperature and was then cooled in ice bath; subsequently, an excess of NaH was centrifuged. The supernatant was poured into a cooled 40 mL volume of phosphate buffer. The mixture was extracted with AcOEt (4x 100 mL). Organic extracts were combined, washed with H
2O, and dried over MgSO
4. Solvents were evaporated under reduced pressure. The crude product was purified via silica gel column chromatography using a gradient of AcOEt in hexane from 0% to 10% as eluting solvent system.
Yield: 883 mg (27%);
TLC (Hexane:AcOEt 2:1)
Rf: 0.55;
1H NMR (600 MHz, CDCl
3) δ: 7.78 (d, 2H,
Harom), 7.41 (d, 12H,
Harom), 7.27 (m, 20H,
Harom), 4.04 (t, 2H, OCH
2CH
2CH
2CH2OSO
2), 3.55 (p, 1H, TrOCH
2CHOCH
2OTr), 3.48 (t, 2H, O
CH2CH
2CH
2CH
2OSO
2), 3.23 (ddd, 4H, TrO
CH2CH), 2.43 (s, 3H,
CH3tosyl), 1.75 (m, 2H, OCH
2CH
2CH2CH
2OSO
2), and 1.58 (m, 2H, OCH
2CH2CH
2CH
2OSO
2);
13C{1H} NMR (125 MHz, CD
3CN) δ: 144.62 (1C,
aromatictosyl), 144.05 (6C,
aromatictrityl), 133.19 (1C,
aromatictosyl), 129.81 (2C,
aromatictosyl), 128.73 (12C,
aromatictrityl), 127.88 (2C,
aromatictosyl), 127.76 (12C,
aromatictrityl), 126.91 (6C,
aromatictrityl), 86.52 (2C, O
C(Ph)3), 78.59 (1C, TrOCH
2CHOCH
2OTr), 70.51 (1C, OCH
2CH
2CH
2CH2OSO
2-), 69.47 (1C, O
CH2CH
2CH
2CH
2OSO
2-), 63.39 (2C, TrO
CH2CH), 26.10 (1C, OCH
2CH
2CH2CH
2OSO
2), 25.86 (1C, OCH
2CH2CH
2CH
2OSO
2), and 21.64 (1C, CH
3tosyl);
ATR-IR (cm−1): 3054, 3018, 2946, 2929, 2869, 1978, 1732, 1596, 1488, 1447, 1352, 1304, 1218, 1187, 1172, 1122, 1094, 1065, 1029, 992, 950, 922, 841, 811, 768, 745, and 699;
UV-Vis λ
max (nm): 198, 229, and 260;
ESI-
MS (
m/
z): found 825.32 [M + Na]
−, 841.29 [M + K]
−, (calc. for C
52H
50O
6S
1 803.01).
Synthesis of 8,8′-bridged {8,8′-O2P(O)S[(CH2)4OCH(CH2OCPh3)2]-3,3′-Co(1,2-C2B9H10)2} HNEt3 (21). [8,8′-O2P(O)SH-3,3′-Co(1,2-C2B9H10)2] HDBU (13) (440 mg, 0.75 mmol) was dissolved in 20 mL of anhydrous acetone; then, anhydrous Et3N (2.15 mL, 15.42 mmol) was added to the resultant solution under stirring at room temperature. The mixture was heated to 60 °C; then, 4-[1,3-bis(trityloxy)propan-2-yl-oxy]butyl-4-methylbenzenesulfonate (20) (901 mg, 1.12 mmol), which was dissolved in 20 mL of anhydrous AcOEt, was added dropwise. After stirring overnight at 60 °C, the mixture was cooled, and the solvents were evaporated under reduced pressure. The residue was dispersed in CH2Cl2, filtered, and the filtrate was loaded into the silica gel column prepared in CH2Cl2. Chromatography was performed using a gradient of CH3OH in CH2Cl2 from 0 to 3% of CH3OH. Yield: 560 mg (64%); TLC (CH3CN:CHCl3 1:2), Rf: 0.60; 1H NMR (600 MHz, CD3CN) δ: 7.41 (d, 12H, Haromatic), 7.31 (m, 18H, Haromatic), 3.70 (s, 4H, CHcarborane), 3.60 (p, 1H, TrOCH2CHOCH2OTr), 3.45 (t, 2H, PSCH2CH2CH2CH2O), 3.19 (ddd, 4H, CHOCH2OTr), 3,10 (q, 6H, HNCH2CH3), 2.85 (dt, 2H, PSCH2CH2CH2CH2O), 1.71 (m, 2H, PSCH2CH2CH2CH2O), 1.61 (m, 2H, PSCH2CH2CH2CH2O), and 1.24 (t, 9H, NCH2CH3); 13C{1H} NMR (125 MHz, CD3CN) δ: 144.73 (6C, aromatictrityl), 129.10 (12C, aromatictrityl), 128.41 (12C, aromatictrityl), 127.60 (6C, aromatictrityl), 86.89 (2C, OC(Ph)3), 78.47 (1C, TrOCH2CHOCH2OTr), 69.92 (1C, PSCH2CH2CH2CH2O), 63.66 (2C, CHOCH2OTr), 47.28 (3C, HNCH2CH3), 47.18 (4C, CHcarborane), 31.89 (1C, PSCH2CH2CH2CH2O), 22.93 (1C, PSCH2CH2CH2CH2O), 13.96 (1C, PSCH2CH2CH2CH2O), and 8.80 (3C, HNCH2CH3); 11B{1H} NMR (120 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8′), −3.04 (s, 2B, B10,10′), −5.67 (s, 4B, B9,9′,12,12′), −8.55 (s, 4B, B4,4′,7,7′), −19.07 (s, 4B, B5,5′,11,11′), and −27.99 (s, 2B, B6,6′); 11B NMR (120 MHz, CD3CN) δ: 23.02 (s, 2B, B8,8′), −2.98 (d, 2B, B10,10′), −5.64 (d, 4B, B9,9′,12,12′), −8.19 (d, 4B, B4,4′,7,7′), −19.05 (d, 4B, B5,5′,11,11′), and −27.99 (d, 2B, B6,6′); 31P{1H}NMR (202 MHz, CD3CN) δ (ppm) 16.49 (s); 31P NMR (202 MHz, CD3CN) δ: 16.48 (t); ATR-IR (cm−1): 3031, 2929, 2870, 2564, 2564, 2161, 1978, 1644, 1595, 1489, 1447, 1322, 1202, 1134, 1096, 1031, 940, 899, 848, 763, 747, and 697; UV-Vis λmax (nm): 196, 233, 299, and 453; MS (ESI) (m/z): found 1064.52 (calc. for C49B18H63O6P1S1Co1 1064.59).
Synthesis of 8,8′-bridged {8,8′-O2P(O)S[(CH2)4OCH(CH2OCPh3)2]-3,3′-Co[1-(CH2)2OH-1,2-C2B9H10)](1′,2′-C2B9H10)} HNEt3 (22) and {8,8′-O2P(O)S[(CH2)4OCH(CH2OCPh3)2]-3,3′-Co[1-(CH2)2OH-1,2-C2B9H10)] [1′-(CH2)2OH-1′,2′-C2B9H10)]} HNEt3 (23). Compound 21 (175 mg, 0.15 mmol) was dried via co-evaporation with anhydrous benzene and then kept under vacuum over P2O5 overnight. Then, it was dissolved in anhydrous DME (3 mL), and the solution was cooled in CO2/isopropanol cooling bath. After 15 min, n-BuLi (140 µL, 1.6 M solution in hexane, 1.5 eq) was added, and the reaction mixture was stirred for 10 min. Afterwards, the cooling bath was removed, and the mixture was stirred for next 10 min. Then, the reaction mixture was cooled again in cooling bath and another portion of n-BuLi (140 µL) was added. After 15 min, ethylene oxide (200 µL, 2.9–3.1 M solution in THF)) was added, and the reaction was left overnight in cooling bath. Then, CH2Cl2 (5 mL) was added to the reaction mixture, the reaction was quenched via addition of water, and then the organic solution was washed three times with 5 mL portions of water. Organic layer was separated and dried over MgSO4; then, solvents were evaporated. Crude product was purified, and mono- and bis-substituted products were separated via silica gel column chromatography using a gradient of MeOH in CH2Cl2 from 0 to 3% of MeOH. (22): Yield: 17 mg (10%); TLC (MeOH:CH2Cl2 1:12.5): Rf: 0.13; 1H NMR (500 MHz, CD3CN): δ (ppm) 7.41 (d, 12H, Haromatic), 7.31 (m, 18H, Haromatic), 3.90 (s, CHcarborane), 3.78 to 3.54 (m, overlapped, CHcarborane, HOCH2CH2Ccarb, TrOCH2CHOCH2OTr), 3.44 (t, 2H, PSCH2CH2CH2CH2O-), 3.19 (ddd, 4H, CHOCH2OTr), 3.02 to 2.64 (m, overlapped, PSCH2CH2CH2CH2O-, HOCH2CH2Ccarb), and 1.65 (m, 4H, overlapped, PSCH2CH2CH2CH2O); 11B{1H} NMR (120 MHz, CD3CN) δ (ppm) 25.37, 24.65, 23.69, 22.93 (in ratio 2:2:1:1) 26.6 to 21.37 (m, overlapped diastereoizomeric B8,8′), 0.52 to −11.61 (m, overlapped diastereoizomeric, B10,10′,9,9′,12,12′,4,4′,7,7′), −12.05 to −11.61 (d, overlapped diastereoizomeric B5,5′,11,11′), and −21.45 to −28.25 (s, overlapped diastereoizomeric B6,6′); 11B NMR (120 MHz, CD3CN) δ (ppm) 27.17 to 21.57 (m, overlapped diastereoizomeric B8,8′), 2.85 to −11.77 (m, overlapped diastereoizomeric, B10,10′,9,9′,12,12′,4,4′,7,7′), −11.82 to −21.00 (d, overlapped diastereoizomeric B5,5′,11,11′), and −21.10 to −29.92 (s, overlapped diastereoizomeric B6,6′); 31P{1H}NMR (202 MHz, CD3CN) δ: 14.94, 14.58, 14.43, 14.12, and 13.49 (in ratio: 4:1:2:1.5:15); 31P NMR (202 MHz, CD3CN) δ: 14.94 (t), 14.42 (t), 14.12 (t), 13.49 (t); ATR-IR (cm−1): 3630, 3370, 3057, 3031, 2925, 2869, 2565, 2161, 1979, 1596, 1489, 1448, 1255, 1202, 1128, 1077, 1032, 985, 898, 871, 763, 746, and 699; ESI-MS (m/z): found: 1108.54 m/z (calc. for C51B18H67O7P1S1Co1 1108.64).
(23): Yield: 21 mg (12%); TLC (MeOH:CH2Cl2 1:12.5): Rf: 0.27; 1H NMR (500 MHz, CD3CN) δ: 7.40 (d, 12H, Haromatic), 7.31 (m, 18H, Haromatic), 3.84 to 3.51 (m, overlapped, CHcarborane, HOCH2CH2-, TrOCH2CHOCH2OTr,), 3.44 (t, 2H, PSCH2CH2CH2CH2O-), 3.18 (ddd, 4H, CHOCH2OTr), 3.12 to 2.66 (m, overlapped, PSCH2CH2CH2CH2O, HOCH2CH2Ccarborane,), and 1.65 to 1.55 (m, 4H, overllaped, PSCH2CH2CH2CH2O); 11B{1H} NMR (120 MHz, CD3CN) δ: 25.36, 24.41, 23.65, 22.80, 22.23 (in ratio 1:2:1.5:1:1), 26.59 to 20.45 (m, overlapped diastereoizomeric B8,8′), 2.75 to −12.40 (m, overlapped diastereoizomeric, B10,10′,9,9′,12,12′,4,4′,7,7′), and −12.34 to −24.64 (m overlapped diastereoizomeric B5,5′,11,11′6,6′); 11B NMR (120 MHz, CD3CN) δ: 26.65 to 20.21 (m, overlapped diastereoizomeric B8,8′), 2.32 to −12.88 (m, overlapped diastereoizomeric, B10,10′,9,9′,12,12′,4,4′,7,7′), −12.84 to −25.84 (m overlapped diastereoizomeric B5,5′,11,11′6,6′); 31P{1H}NMR (202 MHz, CD3CN) δ: 15.00, 14.16, 14.02, 13.38 (in ratio 5:1:3:1); 31P NMR (202 MHz, CD3CN) δ: 15.99 (t), 14.40 (s), 14.01 (t), and 13.38 (t); ATR-IR (cm−1): 3566, 3357, 3056, 3027, 2920, 2889, 2857, 2565, 2166, 1596, 1489, 1448, 1291, 1255, 1201, 1120, 1078, 1032, 1001, 889, 871, 764, 746, and 699 ESI-MS (m/z): found: 1152.57 (calc. for C53B18H71O8P1S1Co 1152.69).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
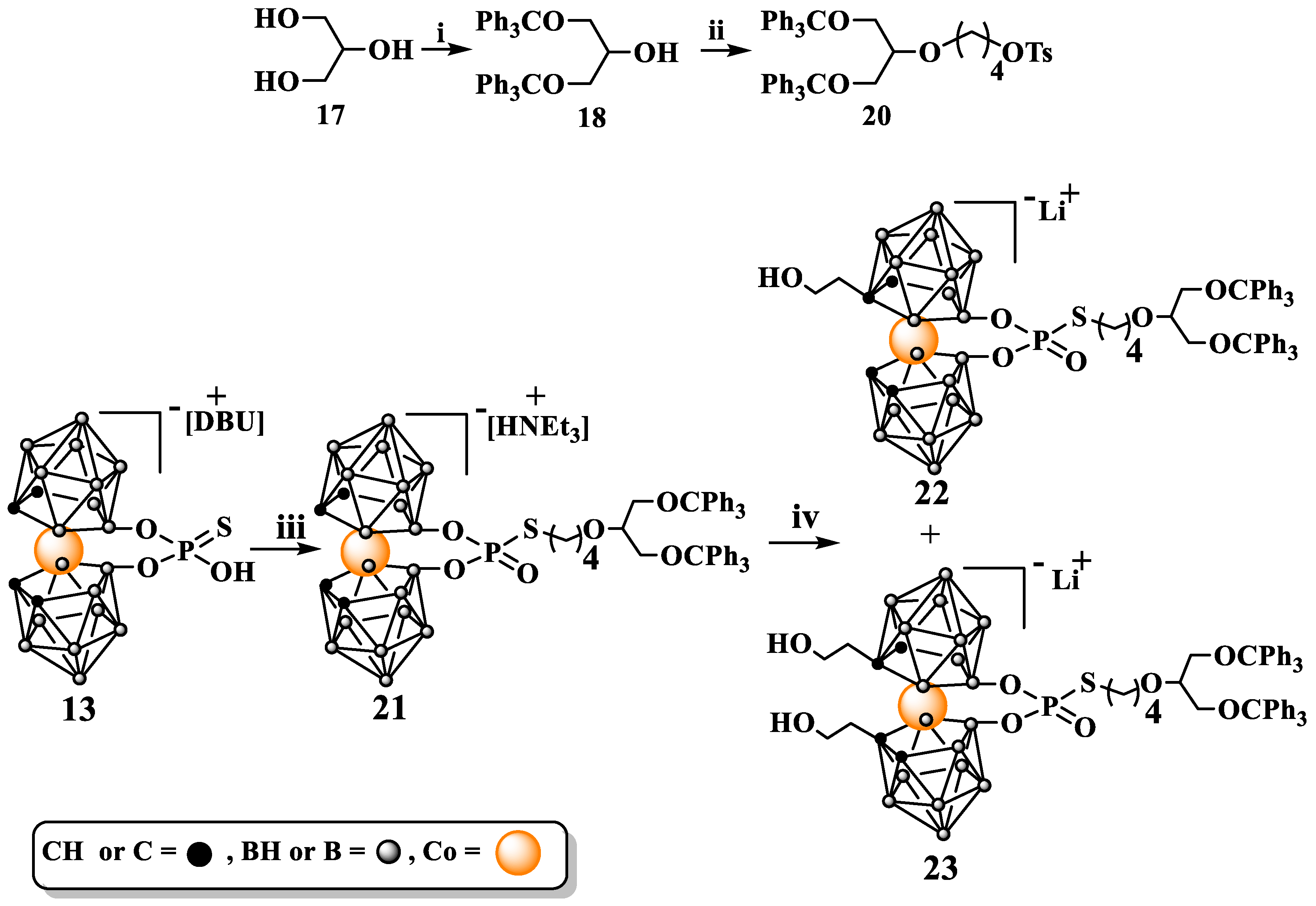{kind=link}






