
One-Pot Solvent-Involved Synthesis of 5-O-Substituted 5H-Chromeno[2,3-b]pyridines
 , ,
, ,  ,
,
Abstract
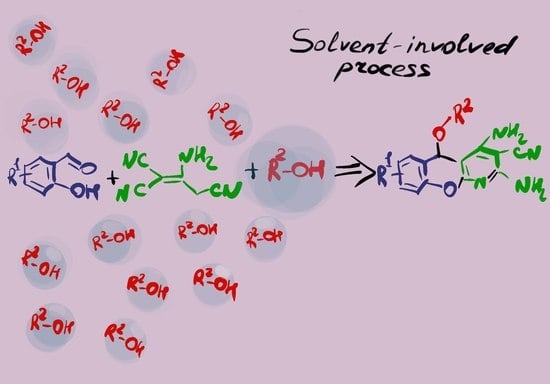
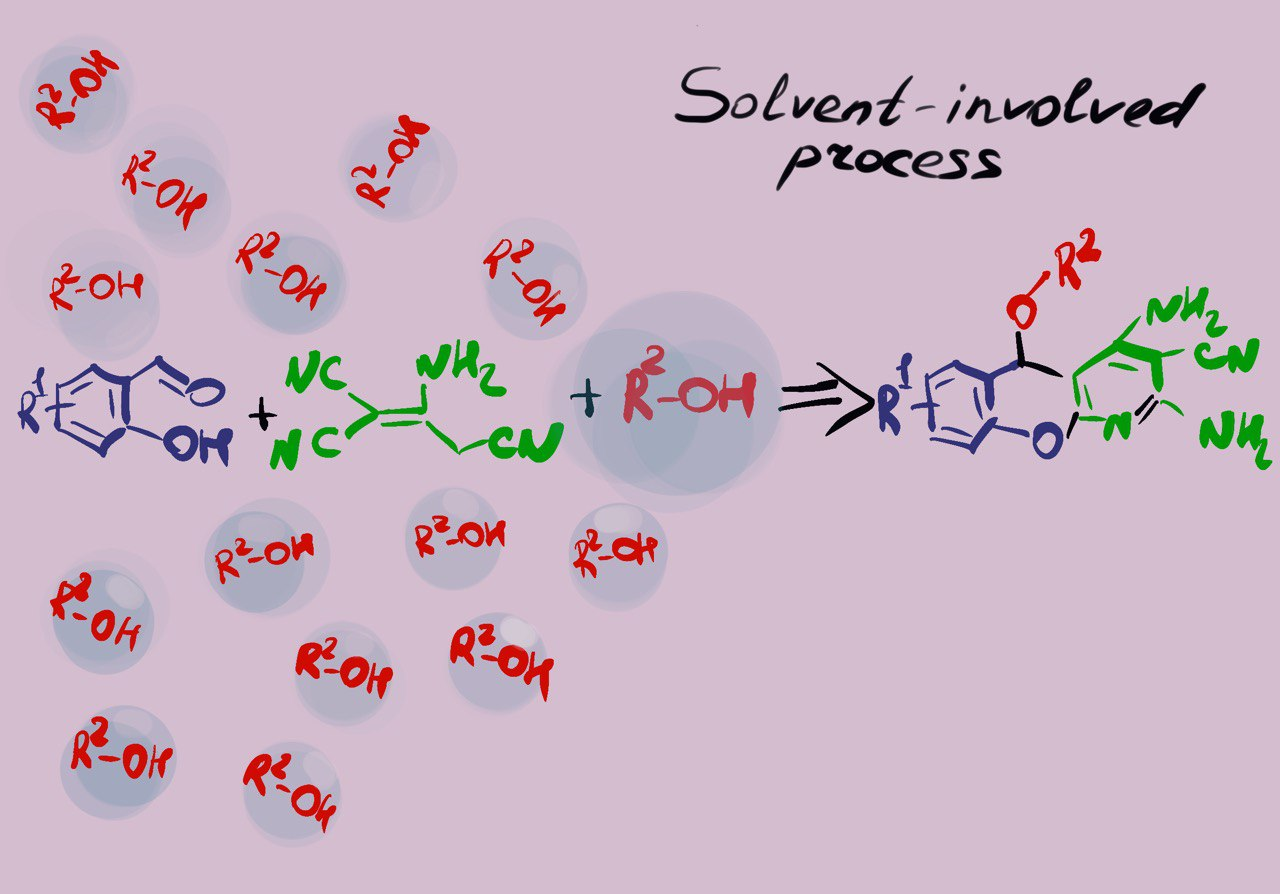
:
1. Introduction
2. Results and Discussion
2.1. Formation of 5-Alkoxy-5H-Chromeno[2,3-b]pyridine 4
2.2. Mechanism of Formation of 5-Methoxy-5H-Chromeno[2,3-b]pyridine 4a

2.3. One-Pot Synthesis of 5-Alkoxy-5H-Chromeno[2,3-b]pyridines 4a–i

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Cat. Amount (mol%) | Solvent Amount st. II (mL) | Time st. II (h) | Temp. (°C) | Yield (%) 2 |
|---|---|---|---|---|---|---|
| 1 | Morpholine | 10 | 20 | 1 | 65 | 80 |
| 2 | Piperidine | 10 | 20 | 1 | 65 | 82 |
| 3 | Pyridine | 10 | 20 | 1 | 65 | 71 |
| 4 | Et3N | 10 | 20 | 1 | 65 | 94 |
| 5 | AcONa | 10 | 20 | 1 | 65 | 48 3 |
| 6 | NaOH | 10 | 20 | 1 | 65 | 52 3 |
| 7 | KF | 10 | 20 | 1 | 65 | 29 3 |
| 8 | Et3N | 20 | 20 | 1 | 65 | 95 |
| 9 | Et3N | 10 | 15 | 1 | 65 | 90 |
| 10 | Et3N | 10 | 25 | 1 | 65 | 82 |
| 11 | Et3N | 10 | 20 | 0.5 | 65 | 93 |
| 12 | Et3N | 10 | 20 | 2 | 65 | 94 |
| 13 | Et3N | 10 | 20 | 0.5 | 23 (rt) | 10 3 |
2.4. 2D-NMR Study of the Structure of Compound 4d
3. Materials and Methods
3.1. General Information
3.2. One-Pot Synthesis of 5-Alkoxy-5H-Chromeno[2,3-b]pyridines 4a–i
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: New York, NY, USA, 1998; p. 30. ISBN 9780198506980. [Google Scholar]
- Anastas, P.; Eghbali, N. Green Chemistry: Principles and Practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Muthengi, A.; Zhang, X.; Dhawan, G.; Zhang, W.; Corsinia, F.; Zhang, W. Sequential (3 + 2) cycloaddition and (5 + n) annulation for modular synthesis of dihydrobenzoxazines, tetrahydrobenzoxazepines and tetrahydrobenzoxazocines. Green Chem. 2018, 20, 3134–3139. [Google Scholar] [CrossRef]
- Zhang, W.; Yi, W.B. Introduction to PASE Synthesis. In Pot, Atom, and Step Economy (PASE) Synthesis; Springer Briefs in Molecular Science; Springer International Publishing: Cham, Switzerland, 2019; pp. 1–4. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Fakhrutdinov, A.N.; Goloveshkin, A.S.; Egorov, M.P. Pot-, Atom- and Step-Economic (PASE) Multicomponent approach to the 5-(Dialkylphosphonate)-Substituted 2,4-Diamino-5H-chromeno[2,3-b]pyridine scaffold. Eur. J. Org. Chem. 2019, 2019, 4171–4178. [Google Scholar] [CrossRef]
- Moseev, T.D.; Varaksin, M.V.; Gorlov, D.A.; Nikiforov, E.A.; Kopchuk, D.S.; Starnovskaya, E.S.; Khasanov, A.F.; Zyryanov, G.V.; Charushin, V.N.; Chupakhin, O.N. Direct CH/CLi coupling of 1,2,4-triazines with C6F5Li followed by aza-Diels-Alder reaction as a pot, atom, and step economy (PASE) approach towards novel fluorinated 2,2′-bipyridine fluorophores. J. Fluor. Chem. 2019, 224, 89–99. [Google Scholar] [CrossRef]
- Romanov, A.R.; Rulev, A.Y.; Ushakov, I.A.; Muzalevskiy, V.M.; Nenajdenko, V.G. One-Pot, Atom and Step Economy (PASE) Assembly of Trifluoromethylated Pyrimidines from CF3-Ynones. Eur. J. Org. Chem. 2017, 2017, 4121–4129. [Google Scholar] [CrossRef]
- Zhang, W.; Yi, W.B. Pot, Atom, and Step Economy (PASE) Synthesis; Springer Briefs in Molecular Science; Springer International Publishing: Cham, Switzerland, 2019; ISBN 9783030225964. [Google Scholar] [CrossRef]
- Clarke, P.A.; Santos, S.; Martin, W.H.C. Combining pot, atom and step economy (PASE) in organic synthesis. Synthesis of tetrahydropyran-4-ones. Green Chem. 2007, 9, 438–440. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, K.; Toda, F. Solvent-Free Organic Synthesis. Chem. Rev. 2000, 100, 1025–1074. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Chowdhury, S. Recent developments in solvent-free multicomponent reactions: A perfect synergy for eco-compatible organic synthesis. RSC Adv. 2012, 2, 4547–4592. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Ryzhkov, F.V.; Anisina, Y.E. ’Solvent-free’ and ’on-solvent’ multicomponent reaction of isatins, malononitrile, and bicyclic CH-acids: Fast and efficient way to medicinal privileged spirooxindole scaffold. Arkivoc 2018, 4, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Banfi, L.; Guanti, G.; Riva, R.; Basso, A. Multicomponent reactions in solid-phase synthesis. Curr. Opin. Drug Discov. Devel. 2007, 10, 704–714. Available online: https://pubmed.ncbi.nlm.nih.gov/17987522/ (accessed on 20 November 2022).
- Cankařová, N.; Krchňák, V. Isocyanide Multicomponent Reactions on Solid Phase: State of the Art and Future Application. Int. J. Mol. Sci. 2020, 21, 9160. [Google Scholar] [CrossRef] [PubMed]
- Elinson, M.N.; Ryzhkov, F.V.; Zaimovskaya, T.A.; Egorov, M.P. Non-catalytic Solvent-Free Synthesis of 5,6,7,8-Tetrahydro-4H-chromenes from Aldehydes, Dimedone and Malononitrile at Ambient Temperature. Mendeleev Commun. 2015, 25, 185–187. [Google Scholar] [CrossRef]
- Kumaravel, K.; Rajarathinam, B.; Vasuki, G. Water-triggered union of multi-component reactions towards the synthesis of a 4H-chromene hybrid scaffold. RSC Adv. 2020, 10, 29109–29113. [Google Scholar] [CrossRef] [PubMed]
- Nasiriani, T.; Javanbakht, S.; Nazeri, M.T.; Farhid, H.; Khodkari, V.; Shaabani, A. Isocyanide-Based Multicomponent Reactions in Water: Advanced Green Tools for the Synthesis of Heterocyclic Compounds. Top. Curr. Chem. (Z) 2022, 380, 50. [Google Scholar] [CrossRef] [PubMed]
- Elinson, M.N.; Nasybullin, R.F.; Ryzhkov, F.V.; Egorov, M.P. Solvent-free and ‘on-water’ multicomponent assembling of salicylaldehydes, malononitrile and 3-methyl-2-pyrazolin-5-one: A fast and efficient route to the 2-amino-4-(1H-pyrazol-4-yl)-4H-chromene scaffold. C. R. Chim. 2014, 17, 437–442. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Sarma, K.D. Multicomponent reactions are accelerated in water. J. Am. Chem. Soc. 2004, 126, 444–445. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Das Sarma, K. Aqueous medium effects on multi-component reactions. Tetrahedron 2005, 61, 11456–11472. [Google Scholar] [CrossRef]
- Hudson, R.F.; Saville, B. Solvent participation in nucleophilic displacement reactions. Part I. General considerations. J. Chem. Soc. 1955, 4114–4121. [Google Scholar] [CrossRef]
- Abdullah Youssef, A.-R. Solution & Bulk Polymerization. 2019. Available online: https://doi.org/10.13140/RG.2.2.16472.96001/2 (accessed on 1 November 2022).
- Weinstein, D.S.; Gong, H.; Doweyko, A.M.; Cunningham, M.; Habte, S.; Wang, J.H.; Holloway, D.A.; Burke, C.; Gao, L.; Guarino, V.; et al. Azaxanthene based selective glucocorticoid receptor modulators: Design, synthesis, and pharmacological evaluation of (S)-4-(5-(1-((1,3,4-thiadiazol-2-yl)amino)-2-methyl-1-oxopropan-2-yl)-5H-chromeno[2,3-b]pyridin-2-yl)-2-fluoro-N,N-dimethylbenzamide (BMS-776532) and its methylene homologue (BMS-791826). J. Med. Chem. 2011, 54, 7318–7833. [Google Scholar] [CrossRef]
- Kolokythas, G.; Pouli, N.; Marakos, P.; Pratsinis, H.; Kletsas, D. Design, synthesis and antiproliferative activity of some new azapyranoxanthenone aminoderivatives. Eur. J. Med. Chem. 2006, 41, 71–79. [Google Scholar] [CrossRef]
- Azuine, M.A.; Tokuda, H.; Takayasu, J.; Enjyo, F.; Mukainaka, T.; Konoshima, T.; Nishino, H.; Kapadia, G. Cancer chemopreventive effect of phenothiazines and related tri-heterocyclic analogues in the 12-O-tetradecanoylphorbol-13-acetate promoted Epstein-Barr virus early antigen activation and the mouse skin two-stage carcinogenesis models. J. Pharmacol. Res. 2004, 49, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, Y.; Goto, K.; Terasawa, M. Method for Treatment of Rheumatism. U.S. Patent 4281001, 6 August 1981. [Google Scholar]
- Ukawa, K.; Ishiguro, T.; Kuriki, H.; Nohara, A. Synthesis of the metabolites and degradation products of 2-amino-7-isopropyl-5-oxo-5H-(1)benzopyrano(2,3-b)pyridine-3-carboxylic acid (Amoxanox). Chem. Pharm. Bull. 1985, 33, 4432–4437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, A.; Tsuruoka, S.; Kanai, Y.; Endou, H.; Saito, K.; Miyamoto, E.; Fujimura, A. Evaluation of the interaction between nonsteroidal anti-inflammatory drugs and methotrexate using human organic anion transporter 3-transfected cells. Eur. J. Pharmacol. 2008, 596, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Oral, E.A.; Reilly, S.M.; Gomez, A.V.; Meral, R.; Butz, L.; Ajluni, N.; Chenevert, T.L.; Korytnaya, E.; Neidert, A.H.; Hench, R.; et al. Inhibition of IKKɛ and TBK1 Improves Glucose Control in a Subset of Patients with Type 2 Diabetes. Cell Metab. 2017, 26, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Ansari, K.R.; Quraishi, M.A.; Singh, A. Chromenopyridin derivatives as environmentally benign corrosion inhibitors for N80 steel in 15% HCl. J. Assoc. Arab. Univ. Basic Appl. Sci. 2017, 22, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Khurana, J.M. An efficient approach for the synthesis of 5-hydroxy-chromeno[2,3-b]pyridines under catalyst and solvent free conditions. Green Chem. 2017, 19, 4153–4156. [Google Scholar] [CrossRef]
- Ghosh, C.K.; Tewari, N.; Morin, C. ChemInform Abstract: Heterocyclic Systems. Part XII. Condensation of 4-OXO-4H-1-BENZOPYRAN-3-CARBONITRILE with Benzoylglycine. Chem. Inf.-Dienst. 1983, 14, 252. [Google Scholar] [CrossRef]
- Ryzhkova, Y.E.; Elinson, M.N.; Maslov, O.I.; Fakhrutdinov, A.N. Multicomponent Synthesis of 2-(2,4-Diamino-3-cyano-5H-chromeno[2,3-b]pyridin-5-yl)malonic Acids in DMSO. Molecules 2021, 26, 6839. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Elinson, M.N.; Anisina, Y.E.; Ryzhkov, F.V.; Goloveshkin, A.S.; Bushmarinov, I.S.; Zlotin, S.G.; Egorov, M.P. Pot, atom and step economic (PASE) synthesis of 5-isoxazolyl-5H-chromeno[2,3-b]pyridine scaffold. Mendeleev Commun. 2015, 25, 424–426. [Google Scholar] [CrossRef]
- Vereshchagin, A.N.; Elinson, M.N.; Anisina, Y.E.; Ryzhkov, F.V.; Novikov, R.A.; Egorov, M.P. PASE Pseudo-Four-Component Synthesis and Docking Studies of New 5-C-Substituted 2,4-Diamino-5H-Chromeno[2,3-b]pyridine-3-Carbonitriles. ChemistrySelect 2017, 2, 4593–4597. [Google Scholar] [CrossRef]
- Elinson, M.N.; Vereshchagin, A.N.; Anisina, Y.E.; Egorov, M.P. Efficient Multicomponent Approach to the Medicinally Relevant 5-aryl-chromeno[2,3-b]pyridine Scaffold. Polycycl. Aromat. Compd. 2020, 40, 108–115. [Google Scholar] [CrossRef]
- Ryzhkova, Y.E.; Ryzhkov, F.V.; Elinson, M.N.; Vereshchagin, A.N.; Egorov, M.P. Pseudo-four-component synthesis of 5-(4-hydroxy-2-oxo-1,2-dihydropyridin-3-yl)-substituted 5H-chromeno[2,3-b]pyridines and estimation of its affinity to sirtuin 2. Arkivoc 2020, 6, 193–208. [Google Scholar] [CrossRef]
- Ryzhkov, F.V.; Ryzhkova, Y.E.; Elinson, M.N.; Vorobyev, S.V.; Fakhrutdinov, A.N.; Vereshchagin, A.N.; Egorov, M.P. Catalyst-Solvent System for PASE Approach to Hydroxyquinolinone-Substituted Chromeno[2,3-b]pyridines Its Quantum Chemical Study and Investigation of Reaction Mechanism. Molecules 2020, 25, 2573. [Google Scholar] [CrossRef] [PubMed]
- Elinson, M.N.; Ryzhkova, Y.E.; Ryzhkov, F.V. Multicomponent design of chromeno[2,3-b]pyridine systems. Russ. Chem. Rev. 2021, 90, 94–115. [Google Scholar] [CrossRef]
- Ali, R.; Mohammad, K.T.; Sobhan, R. A Review on the Syntheses and Applications of the 5H-chromeno[2,3-b]pyridines. Lett. Org. Chem. 2023, 20, 28–53. [Google Scholar] [CrossRef]
- Ryzhkova, Y.E.; Elinson, M.N. 2,4-Diamino-5-(5-amino-3-oxo-2,3-dihydro-1H-pyrazol-4-yl)-5H-chromeno[2,3-b]pyridine-3-carbonitrile. Molbank 2022, 2022, M1399. [Google Scholar] [CrossRef]
- Osyanin, V.A.; Osipov, D.V.; Klimochkin, Y.N. Convenient one-step synthesis of 4-unsubstituted 2-amino-4H-chromene-2-carbonitriles and 5-unsubstituted 5H-chromeno[2,3-b]pyridine-3-carbonitriles from quaternary ammonium salts. Tetrahedron 2012, 68, 5612–5618. [Google Scholar] [CrossRef]
- Kresge, A.J.; Chiang, Y.; Fitzgerald, P.H.; McDonald, R.S.; Schmid, G.H. General acid catalysis in the hydration of simple olefins. Mechanism of olefin hydration. J. Am. Chem. Soc. 1971, 93, 4907–4908. [Google Scholar] [CrossRef]
- Patai, S.; Israeli, Y. 411. The kinetics and mechanisms of carbonyl–methylene condensations. Part VII. The reaction of malononitrile with aromatic aldehydes in ethanol. J. Chem. Soc. 1960, 2025–2030. [Google Scholar] [CrossRef]
- Oliveira-Pinto, S.; Pontes, O.; Lopes, D.; Sampaio-Marques, B.; Costa, M.D.; Carvalho, L.; Gonçalves, C.S.; Costa, B.M.; Maciel, P.; Ludovico, P.; et al. Unravelling the anticancer potential of functionalized chromeno[2,3-b]pyridines for breast cancer treatment. Bioorg. Chem. 2020, 100, 103942. [Google Scholar] [CrossRef]
- Mittelbach, M. An improved and facile synthesis of 2-amino-1,1,3-tricyanopropene. Monatsh. Chem. 1985, 116, 689–691. [Google Scholar] [CrossRef]







 |
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryzhkova, Y.E.; Ryzhkov, F.V.; Elinson, M.N.; Maslov, O.I.; Fakhrutdinov, A.N. One-Pot Solvent-Involved Synthesis of 5-O-Substituted 5H-Chromeno[2,3-b]pyridines. Molecules 2023, 28, 64. https://doi.org/10.3390/molecules28010064
Ryzhkova YE, Ryzhkov FV, Elinson MN, Maslov OI, Fakhrutdinov AN. One-Pot Solvent-Involved Synthesis of 5-O-Substituted 5H-Chromeno[2,3-b]pyridines. Molecules. 2023; 28(1):64. https://doi.org/10.3390/molecules28010064
Chicago/Turabian StyleRyzhkova, Yuliya E., Fedor V. Ryzhkov, Michail N. Elinson, Oleg I. Maslov, and Artem N. Fakhrutdinov. 2023. "One-Pot Solvent-Involved Synthesis of 5-O-Substituted 5H-Chromeno[2,3-b]pyridines" Molecules 28, no. 1: 64. https://doi.org/10.3390/molecules28010064