

Room Temperature Reduction of Titanium Tetrachloride-Activated Nitriles to Primary Amines with Ammonia-Borane
Abstract
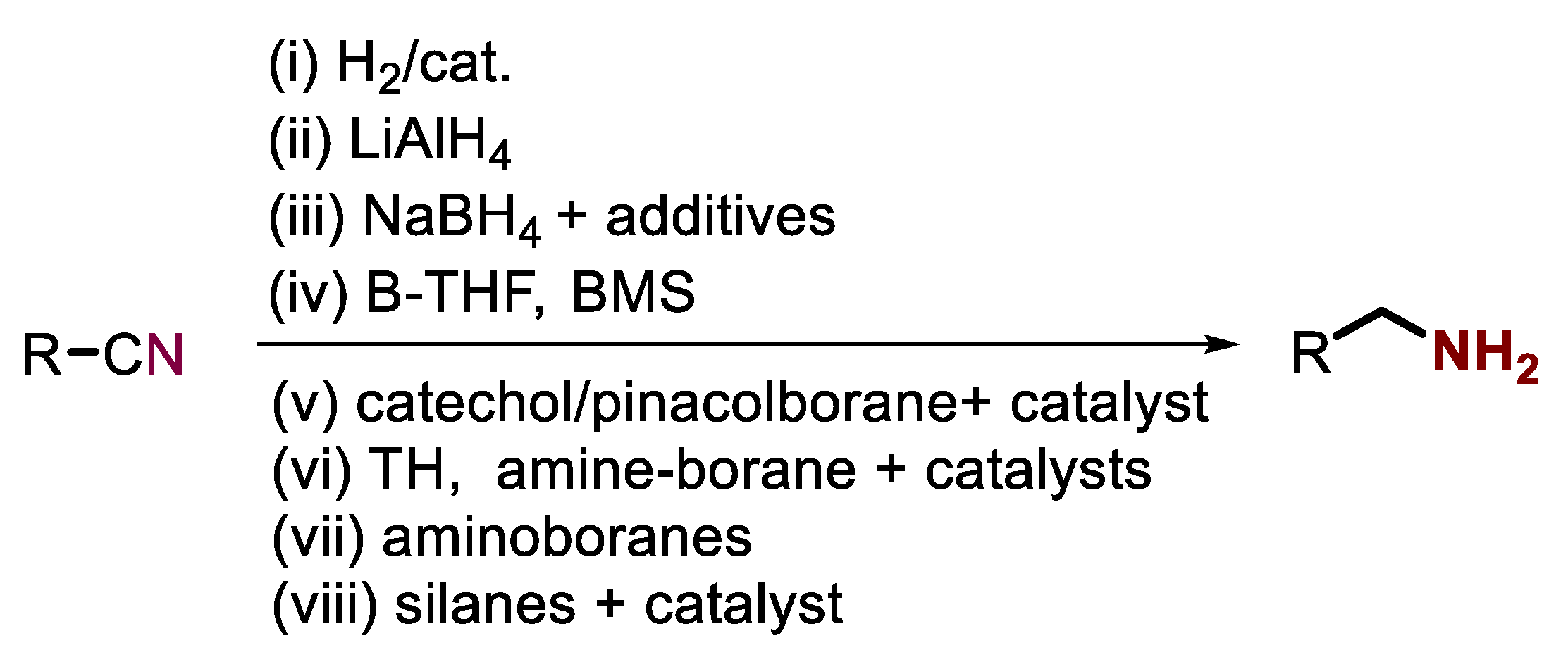
:1. Introduction
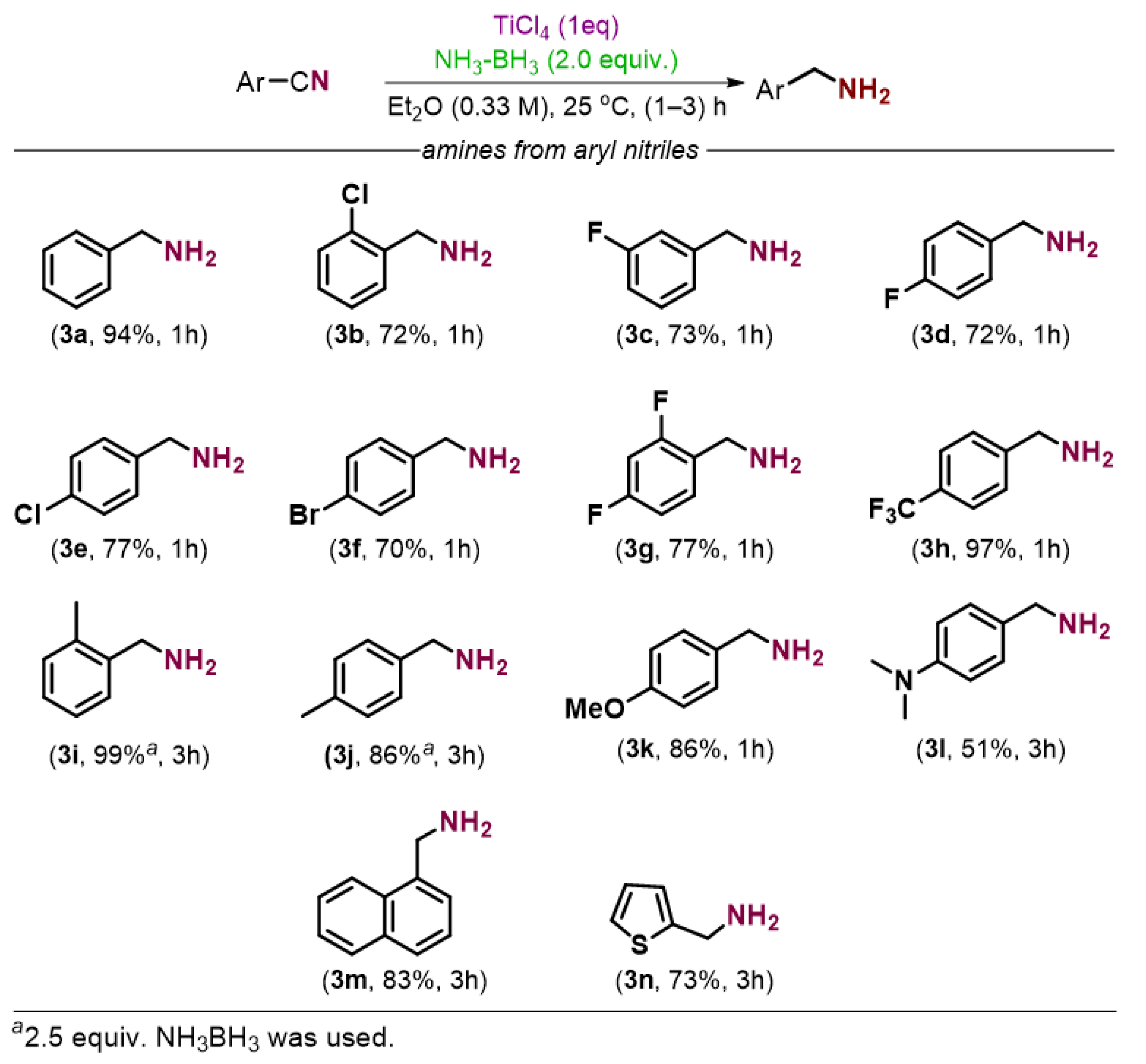
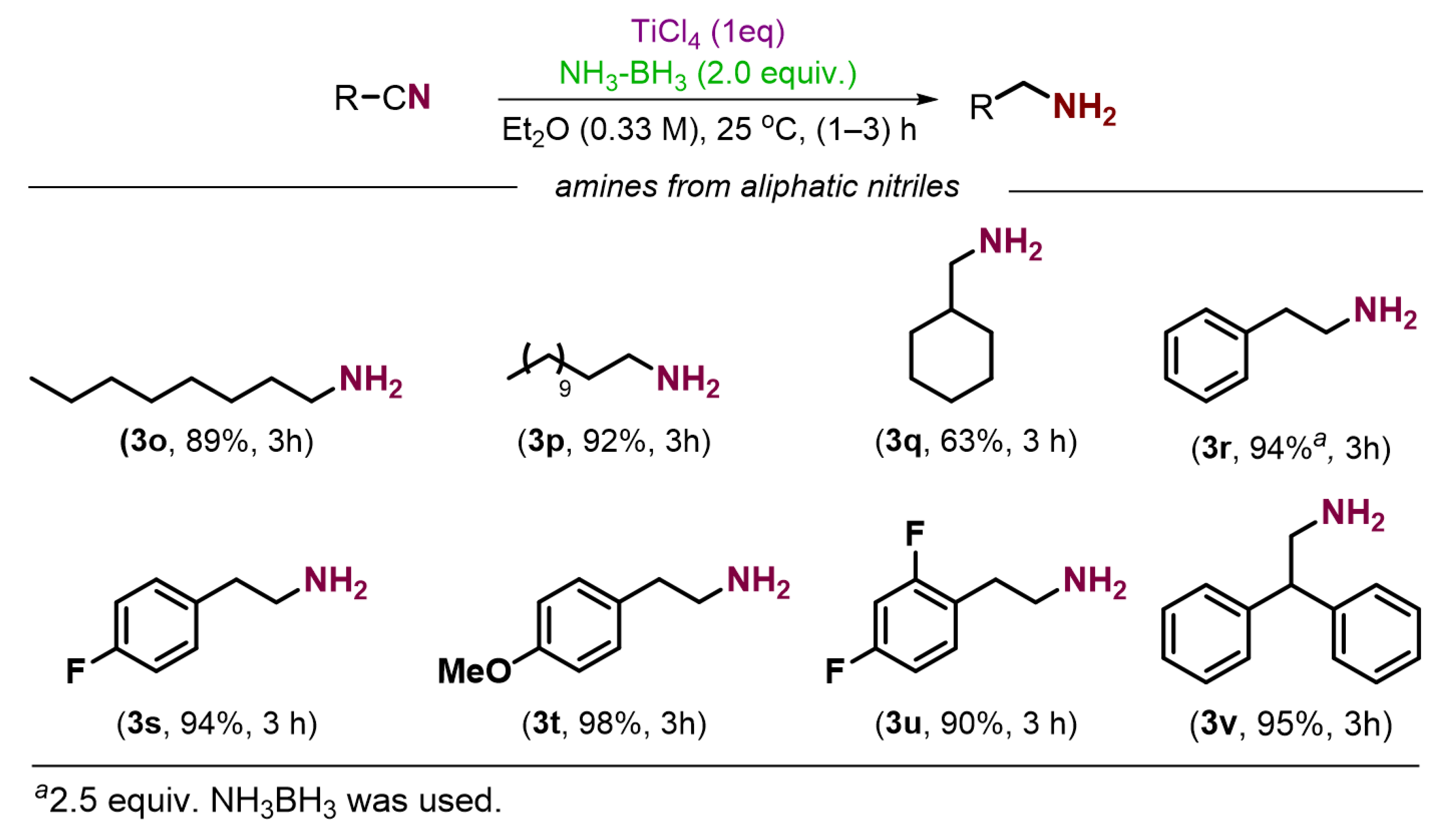
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Experimental
3.2.1. General Procedure for the Preparation of Amines from Nitriles
3.2.2. Characterization of Product Amines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ricci, A. Amino Group Chemistry—From Synthesis to the Life Sciences; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Llevot, A.; Dannecker, P.-K.; von Czapiewski, M.; Over, L.C.; Söyer, Z.; Meier, M.A.R. Renewability is not Enough: Recent Advances in the Sustainable Synthesis of Biomass-Derived Monomers and Polymers. Chem. Eur. J. 2016, 22, 11510–11521. [Google Scholar] [CrossRef] [PubMed]
- Roose, P.; Eller, K.; Henkes, E.; Rossbacher, R.; Höke, H. Amines, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Ricci, A. Modern Amination Methods; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Ricci, A.; Bernardi, L. Methodologies in Amine Synthesis: Challenges and Applications; Wiley-VCH: Weinheim, Germany, 2021. [Google Scholar]
- Afanasyev, O.I.; Kuchuk, E.; Usanov, D.L.; Chusov, D. Reductive Amination in the Synthesis of Pharmaceuticals. Chem. Rev. 2019, 119, 11857–11911. [Google Scholar] [CrossRef] [PubMed]
- Irrgang, T.; Kempe, R. Transition-Metal-Catalyzed Reductive Amination Employing Hydrogen. Chem. Rev. 2020, 120, 9583–9674. [Google Scholar] [CrossRef]
- Abdel-Magid, A.F.; Mehrman, S.J. A Review on the Use of Sodium Triacetoxyborohydride in the Reductive Amination of Ketones and Aldehydes. Org. Process. Res. Dev. 2006, 10, 971–1031. [Google Scholar] [CrossRef]
- Das, S.; Maity, J.; Panda, T.K. Metal/Non-Metal Catalyzed Activation of Organic Nitriles. Chem. Rec. 2022, 22, e202200192. [Google Scholar] [CrossRef]
- Monguchi, Y.; Mizuno, M.; Ichikawa, T.; Fujita, Y.; Murakami, E.; Hattori, T.; Maegawa, T.; Sawama, Y.; Sajiki, H. Catalyst-Dependent Selective Hydrogenation of Nitriles: Selective Synthesis of Tertiary and Secondary Amines. J. Org. Chem. 2017, 82, 10939–10944. [Google Scholar] [CrossRef]
- Novakov, I.A.; Orlinson, B.S.; Mamutova, N.N.; Savel’Ev, E.N.; Potayonkova, E.A.; Pyntya, L.A.; Nakhod, M.A. Reduction of unsaturated adamantyl-containing nitriles with lithium aluminum hydride in 2-methyltetrahydrofuran. Russ. J. Gen. Chem. 2016, 86, 1255–1258. [Google Scholar] [CrossRef]
- Brown, H.C.; Weissman, P.M.; Yoon, N.M. Selective Reductions. IX. Reaction of Lithium Aluminum Hydride with Selected Organic Compounds Containing Representative Functional Groups. J. Am. Chem. Soc. 1966, 88, 1458–1463. [Google Scholar] [CrossRef]
- Chaikin, S.W.; Brown, W.G. Reduction of Aldehydes, Ketones and Acid Chlorides by Sodium Borohydride. J. Am. Chem. Soc. 1949, 71, 122–125. [Google Scholar] [CrossRef]
- Brown, H.C.; Krishnamurthy, S. Forty years of hydride reductions. Tetrahedron 1979, 35, 567–607. [Google Scholar] [CrossRef]
- Brown, H.C.; Ramachandran, P.V. Sixty years of hydride reductions. Reduct. Org. Synth. Recent Adv. Pract. Appl. 1996, 641, 1–30. [Google Scholar]
- Satoh, T.; Suzuki, S.; Suzuki, Y.; Miyaji, Y.; Imai, Z. Reduction of organic compounds with sodium borohydride-transition metal salt systems: Reduction of organic nitrile, nitro and amide compounds to primary amines. Tetrahedron Lett. 1969, 10, 4555–4558. [Google Scholar] [CrossRef]
- Bang, S.; Kim, J.; Jang, B.W.; Kang, S.-G.; Kwak, C.-H. Reduction of nitrile to primary amine using sodium borohydride: Synthesis and protonation of a cis-octahedral macrocyclic nickel(II) complex bearing two 2-aminoethyl pendant arms. Inorg. Chim. Acta 2016, 444, 176–180. [Google Scholar] [CrossRef]
- Caddick, S.; Judd, D.B.; Lewis, A.K.K.; Reich, M.T.; Williams, M.R. A generic approach for the catalytic reduction of nitriles. Tetrahedron 2003, 59, 5417–5423. [Google Scholar] [CrossRef]
- Liu, S.; Yang, Y.; Zhen, X.; Li, J.; He, H.; Feng, J.; Whiting, A. Enhanced reduction of C–N multiple bonds using sodium borohydride and an amorphous nickel catalyst. Org. Biomol. Chem. 2011, 10, 663–670. [Google Scholar] [CrossRef]
- Periasamy, M.; Thirumalaikumar, M. Methods of enhancement of reactivity and selectivity of sodium borohydride for applications in organic synthesis. J. Organomet. Chem. 2000, 609, 137–151. [Google Scholar] [CrossRef]
- Saavedra, J.Z.; Resendez, A.; Rovira, A.; Eagon, S.; Haddenham, D.; Singaram, B. Reaction of InCl3 with Various Reducing Agents: InCl3–NaBH4-Mediated Reduction of Aromatic and Aliphatic Nitriles to Primary Amines. J. Org. Chem. 2011, 77, 221–228. [Google Scholar] [CrossRef]
- Akabori, S.; Takanohashi, Y. Novel borane–selenium complex: Highly selective reduction of tertiary amides and nitriles to the corresponding amines with sodium borohydride–dialkylselenium dibromide. J. Chem. Soc. Perkin Trans. 1 1991, 22, 479–482. [Google Scholar] [CrossRef]
- Brown, H.C.; Choi, Y.M.; Narasimhan, S. Improved procedure for borane-dimethyl sulfide reduction of nitriles. Synthesis 1981, 8, 605–606. [Google Scholar] [CrossRef]
- Fowler, J.S.; MacGregor, R.R.; Ansari, A.N.; Atkins, H.L.; Wolf, A.P. Radiopharmaceuticals. 12. New rapid synthesis of carbon-11-labeled norepinephrine hydrochloride. J. Med. Chem. 1974, 17, 246–248. [Google Scholar] [CrossRef]
- Geri, J.B.; Szymczak, N.K. A Proton-Switchable Bifunctional Ruthenium Complex That Catalyzes Nitrile Hydroboration. J. Am. Chem. Soc. 2015, 137, 12808–12814. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, S.; Zhu, X.; Yuan, Q.; Wei, Y.; Zhou, S.; Mu, X. Well-Defined Amidate-Functionalized N-Heterocyclic Carbene -Supported Rare-Earth Metal Complexes as Catalysts for Efficient Hydroboration of Unactivated Imines and Nitriles. Inorg. Chem. 2018, 57, 15069–15078. [Google Scholar] [CrossRef] [PubMed]
- Kaithal, A.; Chatterjee, B.; Gunanathan, C. Ruthenium-Catalyzed Selective Hydroboration of Nitriles and Imines. J. Org. Chem. 2016, 81, 11153–11161. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, S.; Sankar, R.V.; Gunanathan, C. A Boron-Nitrogen Double Transborylation Strategy for Borane-Catalyzed Hydroboration of Nitriles. J. Org. Chem. 2022, 87, 12386–12396. [Google Scholar] [CrossRef]
- Van der Waals, D.; Pettman, A.; Williams, J.M.J. Copper-catalysed reductive amination of nitriles and organic-group reductions using dimethylamine borane. RSC Adv. 2014, 4, 51845–51849. [Google Scholar] [CrossRef]
- Sarkar, K.; Das, K.; Kundu, A.; Adhikari, D.; Maji, B. Phosphine-Free Manganese Catalyst Enables Selective Transfer Hydrogenation of Nitriles to Primary and Secondary Amines Using Ammonia–Borane. ACS Catal. 2021, 11, 2786–2794. [Google Scholar] [CrossRef]
- Nixon, T.D.; Whittlesey, M.K.; Williams, J.M. Ruthenium-catalysed transfer hydrogenation reactions with dimethylamine borane. Tetrahedron Lett. 2011, 52, 6652–6654. [Google Scholar] [CrossRef]
- Song, H.; Xiao, Y.; Zhang, Z.; Xiong, W.; Wang, R.; Guo, L.; Zhou, T. Switching Selectivity in Copper-Catalyzed Transfer Hydrogenation of Nitriles to Primary Amine-Boranes and Secondary Amines under Mild Conditions. J. Org. Chem. 2021, 87, 790–800. [Google Scholar] [CrossRef]
- Hou, S.-F.; Chen, J.-Y.; Xue, M.; Jia, M.; Zhai, X.; Liao, R.-Z.; Tung, C.-H.; Wang, W. Cooperative Molybdenum-Thiolate Reactivity for Transfer Hydrogenation of Nitriles. ACS Catal. 2019, 10, 380–390. [Google Scholar] [CrossRef]
- Lau, S.; Gasperini, D.; Webster, R.L. Amine–Boranes as Transfer Hydrogenation and Hydrogenation Reagents: A Mechanistic Perspective. Angew. Chem. Int. Ed. 2020, 60, 14272–14294. [Google Scholar] [CrossRef]
- Haddenham, D.; Pasumansky, L.; DeSoto, J.; Eagon, S.; Singaram, B. Reductions of Aliphatic and Aromatic Nitriles to Primary Amines with Diisopropylaminoborane. J. Org. Chem. 2009, 74, 1964–1970. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Fernandes, A.C. A novel efficient and chemoselective method for the reduction of nitriles using the system silane/oxo-rhenium complexes. Tetrahedron 2011, 67, 8183–8186. [Google Scholar] [CrossRef]
- Itazaki, M.; Nakazawa, H. Selective Double Addition Reaction of an E–H Bond (E = Si, B) to a C≡N Triple Bond of Organonitriles. Molecules 2018, 23, 2769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Itazaki, M.; Nakazawa, H. Selective Double Hydrosilylation of Nitriles Catalyzed by an Iron Complex Containing Indium Trihalide. ChemCatChem 2016, 8, 3323–3325. [Google Scholar] [CrossRef]
- Ito, M.; Itazaki, M.; Nakazawa, H. Selective Double Hydroboration and Dihydroborylsilylation of Organonitriles by an Iron–indium Cooperative Catalytic System. Inorg. Chem. 2017, 56, 13709–13714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laval, S.; Dayoub, W.; Favre-Reguillon, A.; Berthod, M.; Demonchaux, P.; Mignani, G.; Lemaire, M. A mild and efficient method for the reduction of nitriles. Tetrahedron Lett. 2009, 50, 7005–7007. [Google Scholar] [CrossRef]
- Sanagawa, A.; Nagashima, H. Hydrosilane Reduction of Nitriles to Primary Amines by Cobalt–Isocyanide Catalysts. Org. Lett. 2018, 21, 287–291. [Google Scholar] [CrossRef]
- Gandhamsetty, N.; Jeong, J.; Park, J.; Park, S.; Chang, S. Boron-Catalyzed Silylative Reduction of Nitriles in Accessing Primary Amines and Imines. J. Org. Chem. 2015, 80, 7281–7287. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Alawaed, A.A.; Hamann, H.J. TiCl4-Catalyzed Hydroboration of Ketones with Ammonia Borane. J. Org. Chem. 2022, 87, 13259–13269. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Alawaed, A.A.; Hamann, H.J. A Safer Reduction of Carboxylic Acids with Titanium Catalysis. Org. Lett. 2022, 24, 8481–8486. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Drolet, M.P.; Kulkarni, A.S. A non-dissociative open-flask hydroboration with ammonia borane: Ready synthesis of ammonia–trialkylboranes and aminodialkylboranes. Chem. Commun. 2016, 52, 11897–11900. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.V.; Drolet, M.P. Direct, high-yielding, one-step synthesis of vic-diols from aryl alkynes. Tetrahedron Lett. 2018, 59, 967–970. [Google Scholar] [CrossRef]
- Hutchins, R.O.; Learn, K.; Nazer, B.; Pytlewski, D.; Pelter, A. Amine boranes as selective reducing and hydroborating agents. A review. Org. Prep. Proced. Int. 1984, 16, 335–372. [Google Scholar] [CrossRef]
- Ramachandran, P.V.; Kulkarni, A.S.; Zhao, Y.; Mei, J. Amine–boranes bearing borane-incompatible functionalities: Application to selective amine protection and surface functionalization. Chem. Commun. 2016, 52, 11885–11888. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.V.; Kulkarni, A.S. Open-Flask Synthesis of Amine–Boranes via Tandem Amine–Ammonium Salt Equilibration–Metathesis. Inorg. Chem. 2015, 54, 5618–5620. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.V.; Kulkarni, A.S. Water-promoted, safe and scalable preparation of ammonia borane. Int. J. Hydrog. Energy 2017, 42, 1451–1455. [Google Scholar] [CrossRef]
- Amberchan, G.; Snelling, R.A.; Moya, E.; Landi, M.; Lutz, K.; Gatihi, R.; Singaram, B. Reaction of Diisobutylaluminum Borohydride, a Binary Hydride, with Selected Organic Compounds Containing Representative Functional Groups. J. Org. Chem. 2021, 86, 6207–6227. [Google Scholar] [CrossRef]
- Ke, D.; Zhou, S. General Construction of Amine via Reduction of N=X (X = C, O, H) Bonds Mediated by Supported Nickel Boride Nanoclusters. Int. J. Mol. Sci. 2022, 23, 9337. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Quan, Z.; Cai, H.; Zhao, Y.; Wang, B. Mild palladium-catalysed highly efficient hydrogenation of C[triple bond, length as m-dash]N, C–NO2, and C[double bond, length as m-dash]O bonds using H2 of 1 atm in H2O. Green Chem. 2019, 21, 830–838. [Google Scholar] [CrossRef]
- Kita, Y.; Kuwabara, M.; Yamadera, S.; Kamata, K.; Hara, M. Effects of ruthenium hydride species on primary amine synthesis by direct amination of alcohols over a heterogeneous Ru catalyst. Chem. Sci. 2020, 11, 9884–9890. [Google Scholar] [CrossRef]
- Liu, Y.; Quan, Z.; He, S.; Zhao, Z.; Wang, J.; Wang, B. Heterogeneous palladium-based catalyst promoted reduction of oximes to amines: Using H2 at 1 atm in H2O under mild conditions. React. Chem. Eng. 2019, 4, 1145–1152. [Google Scholar] [CrossRef]
- Ishitani, H.; Furiya, Y.; Kobayashi, S. Enantioselective Sequential-Flow Synthesis of Baclofen Precursor via Asymmetric 1,4-Addition and Chemoselective Hydrogenation on Platinum/Carbon/Calcium Phosphate Composites. Chem. Asian J. 2020, 15, 1688–1691. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | LA | R3N-BH3 | LA: R3N-BH3 | Solvent | Time, h | Product Yield a, % |
|---|---|---|---|---|---|---|
| 1 | none | 1a | 0:2 | Et2O | 24 | NR b |
| 2 | none | 1a | 0:1 | THF | 20 | 24 c |
| 3 | none | 1a | 0:2 | THF | 20 | 60 c |
| 4 | TiCl4 | 1a | 0.7:2 | Et2O | 3 | 77 |
| 5 d | TiCl4 | 1a | 1:2 | Et2O | 1 | 95 |
| 6 | TiCl4 | 1a | 1:1.5 | Et2O | 24 | 71 |
| 7 | TiCl4 | 1a | 0.7:2 | CH2Cl2 | 3 | 71 |
| 8 | TiCl4 | 1a | 0.7:2 | THF | 3 | 20 |
| 9 | TiCl4 | 1a | 0.7:2 | pentane | 3 | NR b |
| 10 | TiBr4 | 1a | 0.5:2 | Et2O | 3 | 95 |
| 11 | HfCl4 | 1a | 0.7:2 | Et2O | 3 | 63 |
| 12 | BF3.OEt2 | 1a | 0.7:2 | Et2O | 3 | 17 |
| 13 | AlCl3 | 1a | 0.7:2 | Et2O | 3 | 65 |
| 14 | FeCl3 | 1a | 0.7:2 | Et2O | 3 | 55 |
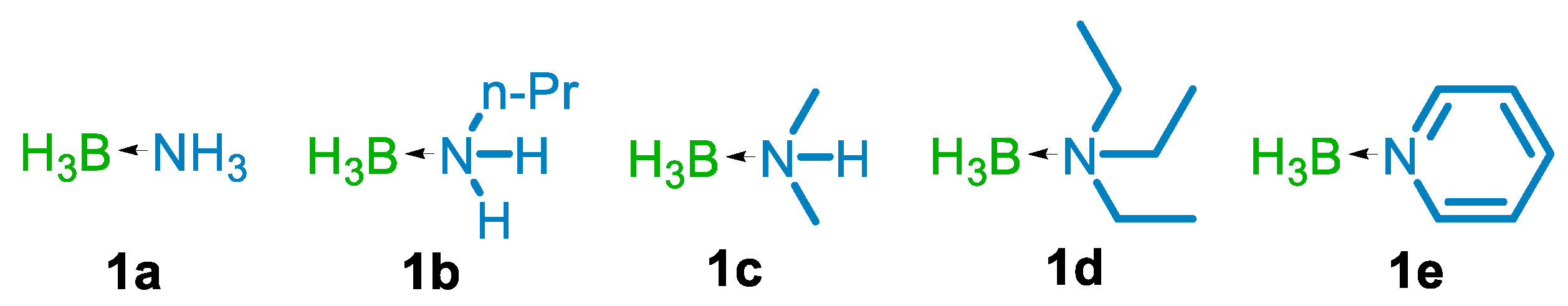
| 15 | TiCl4 | 1b | 1:2 | Et2O | 3 | 16 |
| 16 | TiCl4 | 1c | 1:2 | Et2O | 3 | 17 |
| 17 | TiCl4 | 1d | 1:2 | Et2O | 3 | NR b |
| 18 | TiCl4 | 1e | 1:2 | Et2O | 3 | 34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramachandran, P.V.; Alawaed, A.A. Room Temperature Reduction of Titanium Tetrachloride-Activated Nitriles to Primary Amines with Ammonia-Borane. Molecules 2023, 28, 60. https://doi.org/10.3390/molecules28010060
Ramachandran PV, Alawaed AA. Room Temperature Reduction of Titanium Tetrachloride-Activated Nitriles to Primary Amines with Ammonia-Borane. Molecules. 2023; 28(1):60. https://doi.org/10.3390/molecules28010060
Chicago/Turabian StyleRamachandran, P. Veeraraghavan, and Abdulkhaliq A. Alawaed. 2023. "Room Temperature Reduction of Titanium Tetrachloride-Activated Nitriles to Primary Amines with Ammonia-Borane" Molecules 28, no. 1: 60. https://doi.org/10.3390/molecules28010060