
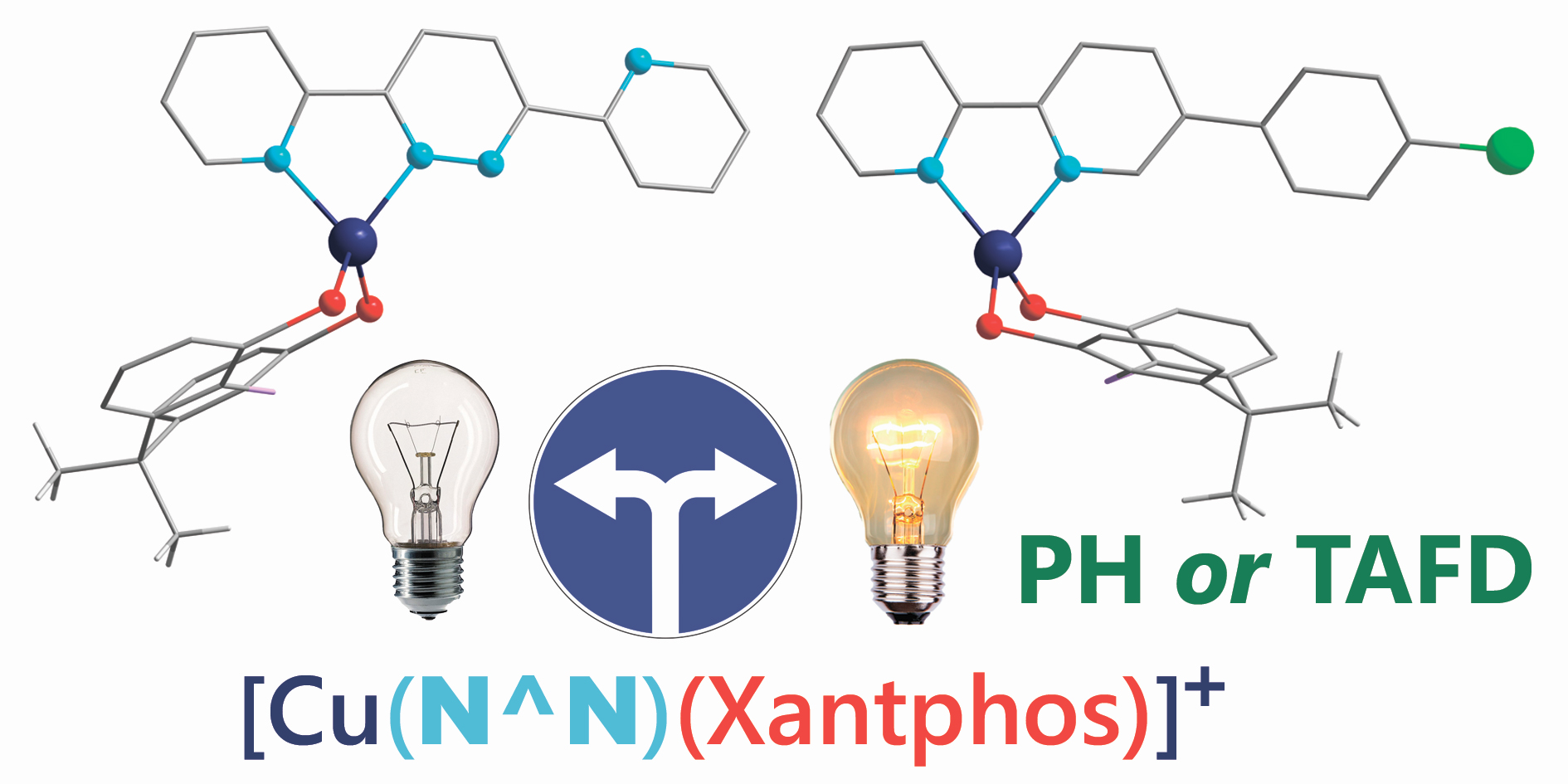
The Tail Wags the Dog: The Far Periphery of the Coordination Environment Manipulates the Photophysical Properties of Heteroleptic Cu(I) Complexes

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
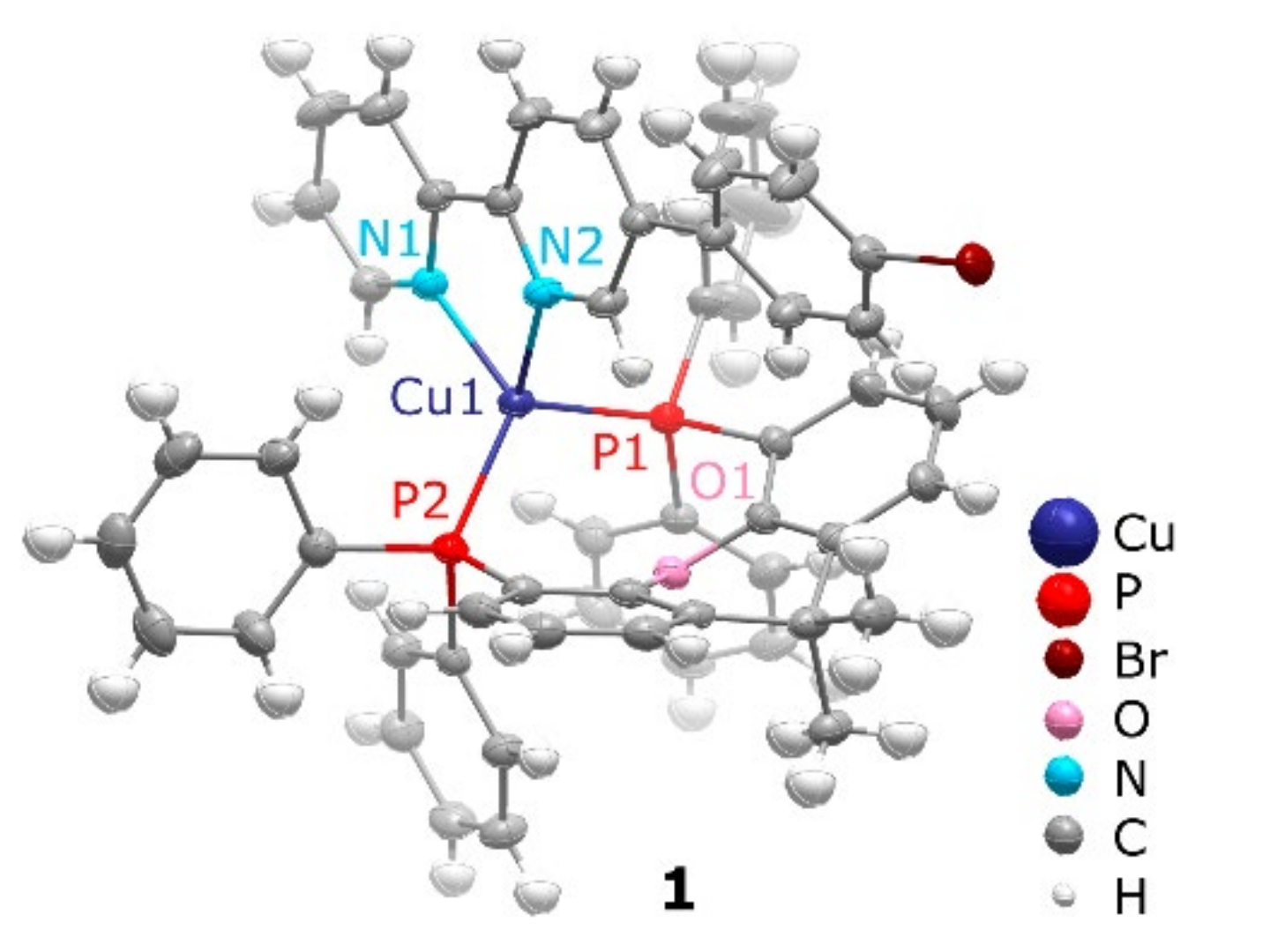
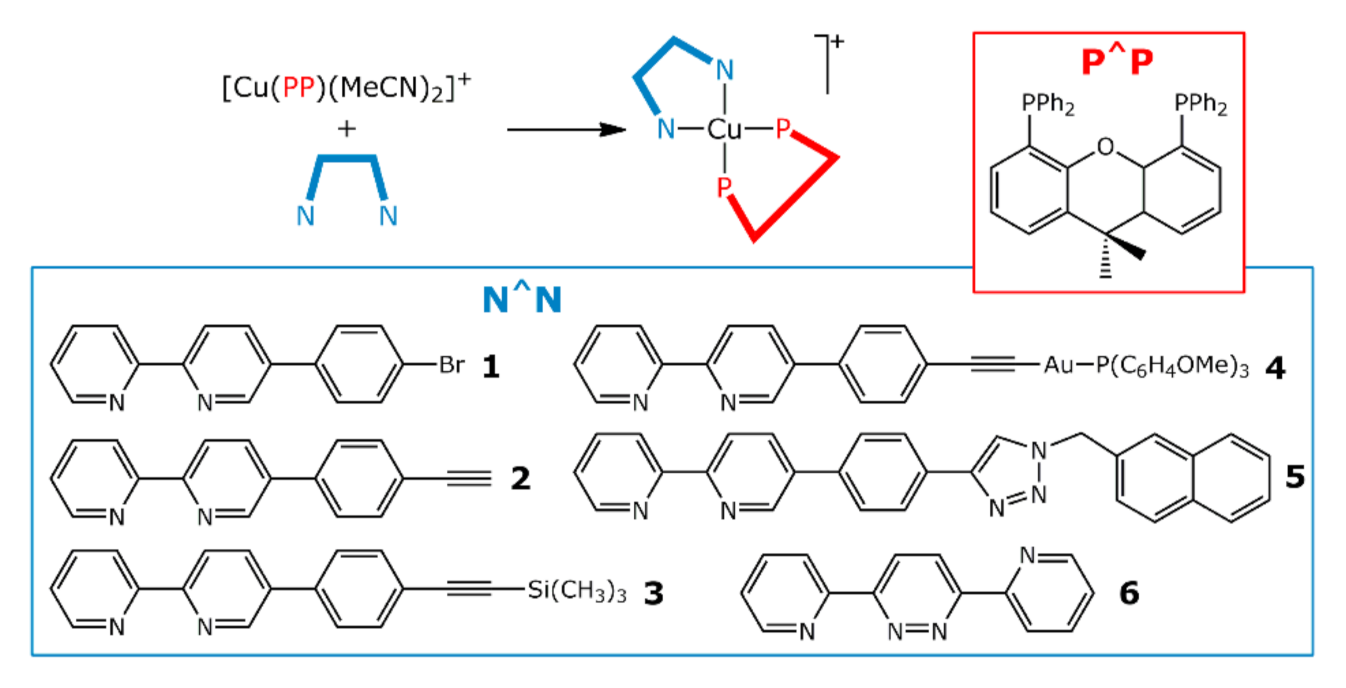
2.1. Synthesis of Cu(I) Complexes
2.2. Electrochemical Properties of 1−6
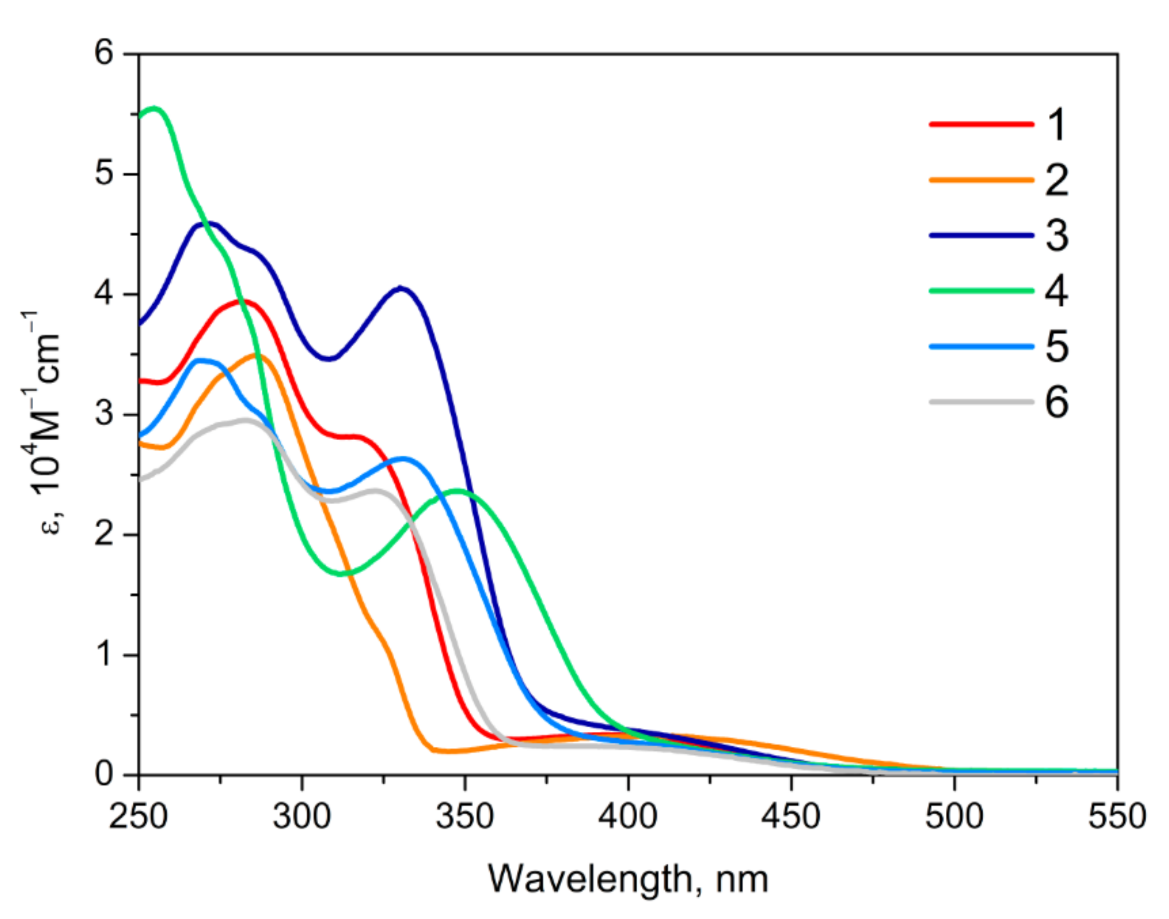
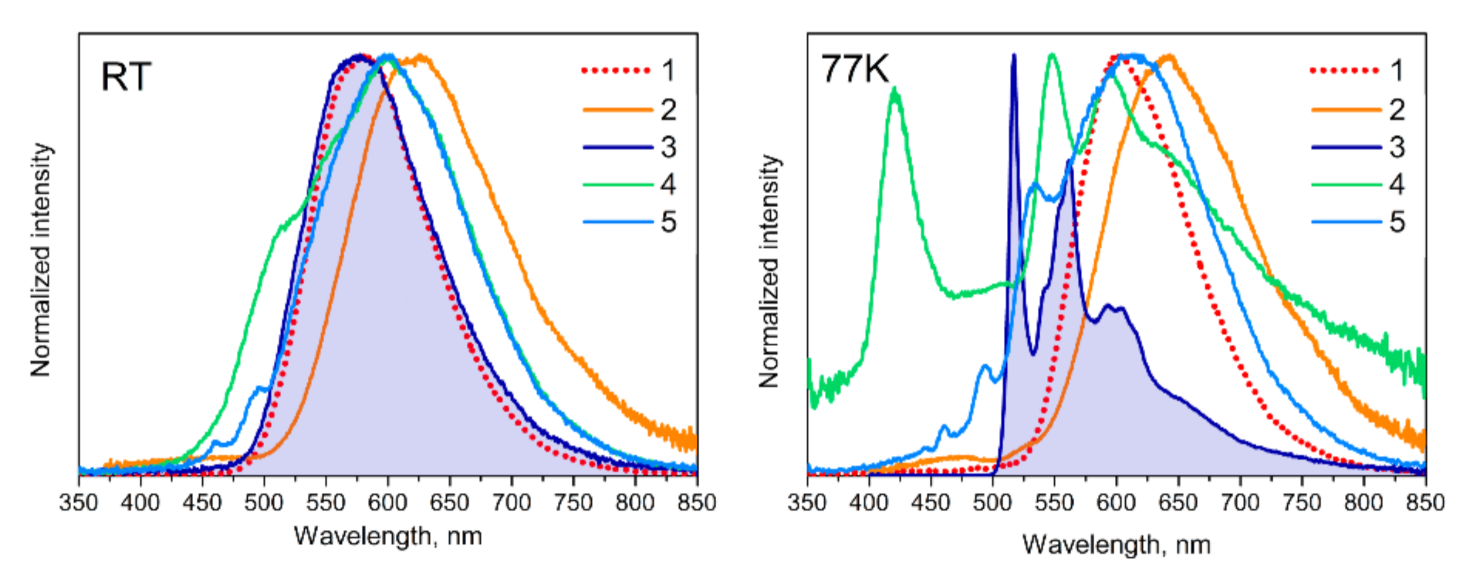
2.3. Photophysical Properties of 1−6
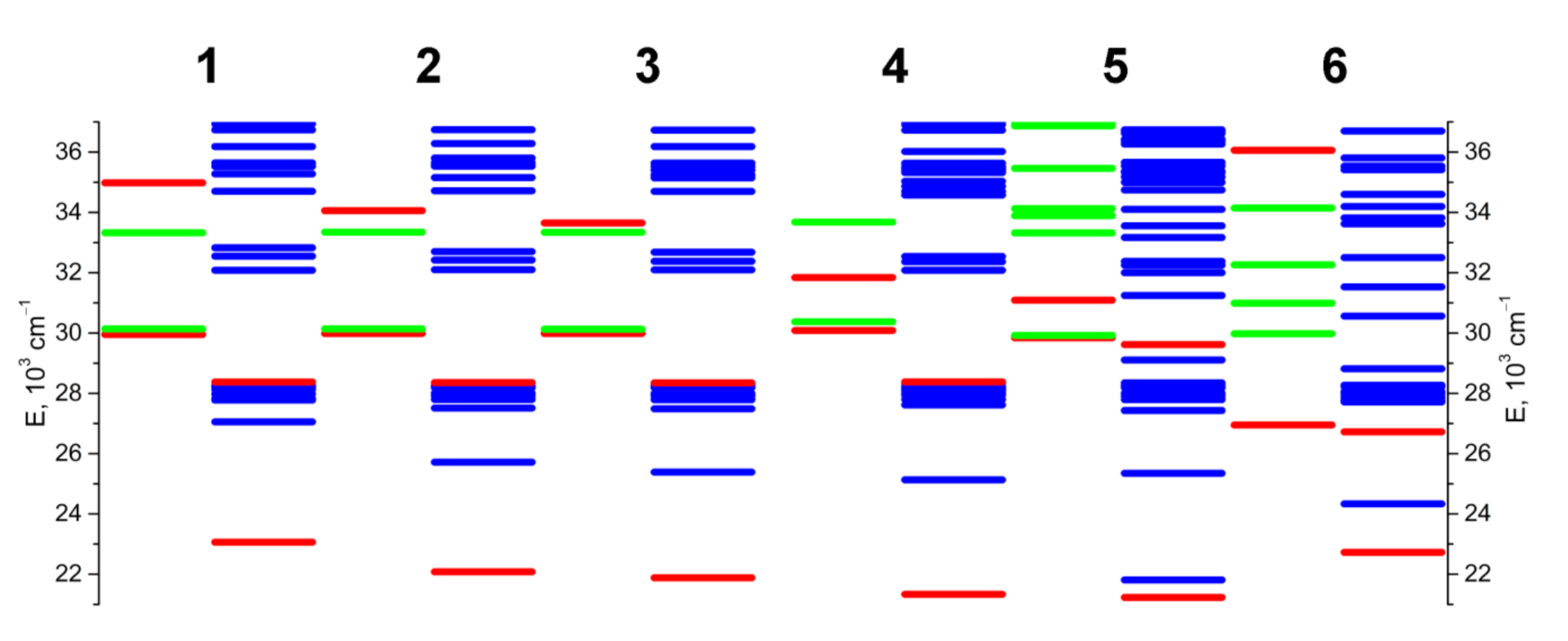
2.4. DFT Calculations
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Zhang, Z.; Fan, J.; Du, J.; Peng, X. Two-channel responsive luminescent chemosensors for dioxygen species: Molecular oxygen, singlet oxygen and superoxide anion. Coord. Chem. Rev. 2021, 427, 213575. [Google Scholar] [CrossRef]
- Jin, H.; Jiang, X.; Sun, Z.; Gui, R. Phosphorescence-based ratiometric probes: Design, preparation and applications in sensing, imaging and biomedicine therapy. Coord. Chem. Rev. 2021, 431, 213694. [Google Scholar] [CrossRef]
- Hong, G.; Gan, X.; Leonhardt, C.; Zhang, Z.; Seibert, J.; Busch, J.M.; Bräse, S. A Brief History of OLEDs-Emitter Development and Industry Milestones. Adv. Mater. 2021, 33, 2005630. [Google Scholar] [CrossRef] [PubMed]
- Twilton, J.; Le, C.; Zhang, P.; Shaw, M.H.; Evans, R.W.; MacMillan, D.W.C. The merger of transition metal and photocatalysis. Nat. Rev. Chem. 2017, 1, 0052. [Google Scholar] [CrossRef]
- Li, J.; Chen, T. Transition metal complexes as photosensitizers for integrated cancer theranostic applications. Coord. Chem. Rev. 2020, 418, 213355. [Google Scholar] [CrossRef]
- Yu, Z.; Hagfeldt, A.; Sun, L. The application of transition metal complexes in hole-transporting layers for perovskite solar cells: Recent progress and future perspectives. Coord. Chem. Rev. 2020, 406, 213143. [Google Scholar] [CrossRef]
- To, W.-P.; Wan, Q.; Tong, G.S.M.; Che, C.-M. Recent Advances in Metal Triplet Emitters with d6, d8, and d10 Electronic Configurations. Trends Chem. 2020, 2, 796–812. [Google Scholar] [CrossRef]
- Li, K.; Chen, Y.; Wang, J.; Yang, C. Diverse emission properties of transition metal complexes beyond exclusive single phosphorescence and their wide applications. Coord. Chem. Rev. 2021, 433, 213755. [Google Scholar] [CrossRef]
- Kritchenkov, I.S.; Zhukovsky, D.D.; Mohamed, A.; Korzhikov-Vlakh, V.A.; Tennikova, T.B.; Lavrentieva, A.; Scheper, T.; Pavlovskiy, V.V.; Porsev, V.V.; Evarestov, R.A.; et al. Functionalized Pt(II) and Ir(III) NIR Emitters and Their Covalent Conjugates with Polymer-Based Nanocarriers. Bioconjug. Chem. 2020, 31, 1327–1343. [Google Scholar] [CrossRef]
- Baranovskii, E.M.; Khistiaeva, V.V.; Deriabin, K.V.; Petrovskii, S.K.; Koshevoy, I.O.; Kolesnikov, I.E.; Grachova, E.V.; Islamova, R.M. Re(I) Complexes as Backbone Substituents and Cross-Linking Agents for Hybrid Luminescent Polysiloxanes and Silicone Rubbers. Molecules 2021, 26, 6866. [Google Scholar] [CrossRef]
- Komarnicka, U.K.; Starosta, R.; Płotek, M.; de Almeida, R.F.M.; Jeżowska-Bojczuk, M.; Kyzioł, A. Copper(I) complexes with phosphine derived from sparfloxacin. Part II: A first insight into the cytotoxic action mode. Dalt. Trans. 2016, 45, 5052–5063. [Google Scholar] [CrossRef] [PubMed]
- Mahoro, G.U.; Fernandez-Cestau, J.; Renaud, J.; Coto, P.B.; Costa, R.D.; Gaillard, S. Recent Advances in Solid-State Lighting Devices Using Transition Metal Complexes Exhibiting Thermally Activated Delayed Fluorescent Emission Mechanism. Adv. Opt. Mater. 2020, 8, 2000260. [Google Scholar] [CrossRef]
- Zhang, Y.; Schulz, M.; Wächtler, M.; Karnahl, M.; Dietzek, B. Heteroleptic diimine–diphosphine Cu(I) complexes as an alternative towards noble-metal based photosensitizers: Design strategies, photophysical properties and perspective applications. Coord. Chem. Rev. 2018, 356, 127–146. [Google Scholar] [CrossRef]
- Minozzi, C.; Caron, A.; Grenier-Petel, J.-C.; Santandrea, J.; Collins, S.K. Heteroleptic Copper(I)-Based Complexes for Photocatalysis: Combinatorial Assembly, Discovery, and Optimization. Angew. Chem. Int. Ed. 2018, 57, 5477–5481. [Google Scholar] [CrossRef] [PubMed]
- Noirbent, G.; Dumur, F. Recent Advances on Copper Complexes as Visible Light Photoinitiators and (Photo) Redox Initiators of Polymerization. Catalysts 2020, 10, 953. [Google Scholar] [CrossRef]
- Leitl, M.J.; Zink, D.M.; Schinabeck, A.; Baumann, T.; Volz, D.; Yersin, H. Copper(I) Complexes for Thermally Activated Delayed Fluorescence: From Photophysical to Device Properties. Top. Curr. Chem. 2016, 374, 25. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Leitl, M.J.; Homeier, H.H.H.; Yersin, H. Cu(I) complexes—Thermally activated delayed fluorescence. Photophysical approach and material design. Coord. Chem. Rev. 2016, 325, 2–28. [Google Scholar] [CrossRef]
- Ravaro, L.P.; Zanoni, K.P.S.; de Camargo, A.S.S. Luminescent Copper(I) complexes as promising materials for the next generation of energy-saving OLED devices. Energy Rep. 2020, 6, 37–45. [Google Scholar] [CrossRef]
- Alkan-Zambada, M.; Hu, X. Cu-Catalyzed Photoredox Chlorosulfonation of Alkenes and Alkynes. J. Org. Chem. 2019, 84, 4525–4533. [Google Scholar] [CrossRef]
- Dierkes, P.; van Leeuwen, P.W.N.M. The bite angle makes the difference: A practical ligand parameter for diphosphine ligands. J. Chem. Soc. Dalt. Trans. 1999, 1519–1530. [Google Scholar] [CrossRef]
- Alkan-Zambada, M.; Constable, E.C.; Housecroft, C.E. The Role of Percent Volume Buried in the Characterization of Copper(I) Complexes for Lighting Purposes. Molecules 2020, 25, 2647. [Google Scholar] [CrossRef]
- McCullough, B.J.; Neyhouse, B.J.; Schrage, B.R.; Reed, D.T.; Osinski, A.J.; Ziegler, C.J.; White, T.A. Visible-Light-Driven Photosystems Using Heteroleptic Cu(I) Photosensitizers and Rh(III) Catalysts To Produce H 2. Inorg. Chem. 2018, 57, 2865–2875. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Tomé, I.; Fyson, J.; Baiao Dias, F.; Monkman, A.P.; Iacobellis, G.; Coppo, P. Copper(i) complexes with bipyridyl and phosphine ligands: A systematic study. Dalt. Trans. 2012, 41, 8669. [Google Scholar] [CrossRef]
- Keller, S.; Brunner, F.; Junquera-Hernández, J.M.; Pertegás, A.; La-Placa, M.-G.; Prescimone, A.; Constable, E.C.; Bolink, H.J.; Ortí, E.; Housecroft, C.E. CF 3 Substitution of [Cu(P^P)(bpy)][PF6] Complexes: Effects on Photophysical Properties and Light-Emitting Electrochemical Cell Performance. Chempluschem 2018, 83, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkan-Zambada, M.; Keller, S.; Martínez-Sarti, L.; Prescimone, A.; Junquera-Hernández, J.M.; Constable, E.C.; Bolink, H.J.; Sessolo, M.; Ortí, E.; Housecroft, C.E. [Cu(P^P)(N^N)][PF6] compounds with bis(phosphane) and 6-alkoxy, 6-alkylthio, 6-phenyloxy and 6-phenylthio-substituted 2,2′-bipyridine ligands for light-emitting electrochemical cells. J. Mater. Chem. C 2018, 6, 8460–8471. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Prescimone, A.; La Placa, M.-G.; Junquera-Hernández, J.M.; Bolink, H.J.; Constable, E.C.; Sessolo, M.; Ortí, E.; Housecroft, C.E. The shiny side of copper: Bringing copper(i) light-emitting electrochemical cells closer to application. RSC Adv. 2020, 10, 22631–22644. [Google Scholar] [CrossRef]
- Mazzeo, F.; Brunner, F.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Intra-Cation versus Inter-Cation π-Contacts in [Cu(P^P)(N^N)][PF6] Complexes. Crystals 2019, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-M.; Ju, P.; Jing, M.-H.; Yu, P.; Huang, Q. Synthesis and Characterization of Three New Emissive Mononuclear CuI Heteroleptic Complexes with Functionalized 6-Cyano-2,2′-bipyridine Chelating Ligands. Aust. J. Chem. 2020, 73, 640. [Google Scholar] [CrossRef]
- Meyer, M.; Brunner, F.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Chimera Diimine Ligands in Emissive [Cu(P^P)(N^N)][PF6] Complexes. Inorganics 2020, 8, 33. [Google Scholar] [CrossRef]
- Brunner, F.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Positional Isomerism in the N^N Ligand: How Much Difference Does a Methyl Group Make in [Cu(P^P)(N^N)]+ Complexes? Molecules 2020, 25, 2760. [Google Scholar] [CrossRef]
- Keller, S.; Alkan-Zambada, M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Extended π-Systems in Diimine Ligands in [Cu(P^P)(N^N)][PF6] Complexes: From 2,2′-Bipyridine to 2-(Pyridin-2-yl)Quinoline. Crystals 2020, 10, 255. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.-Y.; Xin, X.-L.; Guo, Y.-M.; Chen, L.-L.; Liang, Y.-Y.; Xu, M.; Li, X.-L. Synthesis, structure and solid luminescence of copper(I)–bromodiimine–diphosphine complexes. Polyhedron 2015, 101, 23–28. [Google Scholar] [CrossRef]
- Brunner, F.; Martínez-Sarti, L.; Keller, S.; Pertegás, A.; Prescimone, A.; Constable, E.C.; Bolink, H.J.; Housecroft, C.E. Peripheral halo-functionalization in [Cu(N^N)(P^P)]+ emitters: Influence on the performances of light-emitting electrochemical cells. Dalt. Trans. 2016, 45, 15180–15192. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Pertegás, A.; Longo, G.; Martínez, L.; Cerdá, J.; Junquera-Hernández, J.M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E.; Ortí, E.; et al. Shine bright or live long: Substituent effects in [Cu(N^N)(P^P)]+ -based light-emitting electrochemical cells where N^N is a 6-substituted 2,2′-bipyridine. J. Mater. Chem. C 2016, 4, 3857–3871. [Google Scholar] [CrossRef] [Green Version]
- Weber, M.D.; Viciano-Chumillas, M.; Armentano, D.; Cano, J.; Costa, R.D. σ-Hammett parameter: A strategy to enhance both photo- and electro-luminescence features of heteroleptic copper(i) complexes. Dalt. Trans. 2017, 46, 6312–6323. [Google Scholar] [CrossRef]
- Zhang, Y.-R.; Yu, X.; Lin, S.; Jin, Q.-H.; Yang, Y.-P.; Liu, M.; Li, Z.-F.; Zhang, C.-L.; Xin, X.-L. Seven copper (I) complexes of diphosphine ligands and N^N ligands: Syntheses, structural characterizations and spectroscopic properties. Polyhedron 2017, 138, 46–56. [Google Scholar] [CrossRef]
- Brunner, F.; Graber, S.; Baumgartner, Y.; Häussinger, D.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. The effects of introducing sterically demanding aryl substituents in [Cu(N^N)(P^P)]+ complexes. Dalt. Trans. 2017, 46, 6379–6391. [Google Scholar] [CrossRef] [Green Version]
- Keller, S.; Prescimone, A.; Bolink, H.; Sessolo, M.; Longo, G.; Martínez-Sarti, L.; Junquera-Hernández, J.M.; Constable, E.C.; Ortí, E.; Housecroft, C.E. Luminescent copper(i) complexes with bisphosphane and halogen-substituted 2,2′-bipyridine ligands. Dalt. Trans. 2018, 47, 14263–14276. [Google Scholar] [CrossRef]
- Alkan-Zambada, M.; Hu, X. Cu Photoredox Catalysts Supported by a 4,6-Disubstituted 2,2′-Bipyridine Ligand: Application in Chlorotrifluoromethylation of Alkenes. Organometallics 2018, 37, 3928–3935. [Google Scholar] [CrossRef] [Green Version]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; van Rijn, J.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen–sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalt. Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Yang, L.; Powell, D.R.; Houser, R.P. Structural variation in copper(I) complexes with pyridylmethylamide ligands: Structural analysis with a new four-coordinate geometry index, τ4. Dalt. Trans. 2007, 955–964. [Google Scholar] [CrossRef]
- Brunner, F.; Babaei, A.; Pertegás, A.; Junquera-Hernández, J.M.; Prescimone, A.; Constable, E.C.; Bolink, H.J.; Sessolo, M.; Ortí, E.; Housecroft, C.E. Phosphane tuning in heteroleptic [Cu(N^N)(P^P)]+ complexes for light-emitting electrochemical cells. Dalt. Trans. 2019, 48, 446–460. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [Green Version]
- Tárkányi, G.; Király, P.; Pálinkás, G.; Deák, A. Conformational analysis of a helically distorted gold(I) macrocycle derived from xantphos: Evidence for the aurophilic Au…Au interaction from NMR. Magn. Reson. Chem. 2007, 45, 917–924. [Google Scholar] [CrossRef]
- Keller, S.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Copper(i) and silver(i) complexes of 9,9-dimethyl-4,5-bis(di-tert-butylphosphino)xanthene: Photophysical properties and structural rigidity under pressure. Photochem. Photobiol. Sci. 2018, 17, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Mardegan, L.; Tordera, D.; Prescimone, A.; Sessolo, M.; Bolink, H.J.; Constable, E.C.; Housecroft, C.E. A counterion study of a series of [Cu(P^P)(N^N)][A] compounds with bis(phosphane) and 6-methyl and 6,6′-dimethyl-substituted 2,2′-bipyridine ligands for light-emitting electrochemical cells. Dalt. Trans. 2021, 50, 17920–17934. [Google Scholar] [CrossRef]
- Favarin, L.R.V.; Rosa, P.P.; Pizzuti, L.; Machulek, A.; Caires, A.R.L.; Bezerra, L.S.; Pinto, L.M.C.; Maia, G.; Gatto, C.C.; Back, D.F.; et al. Synthesis and structural characterization of new heteroleptic copper(I) complexes based on mixed phosphine/thiocarbamoyl-pyrazoline ligands. Polyhedron 2017, 121, 185–190. [Google Scholar] [CrossRef]
- Yoshida, M.; Yanagida, S.; Saito, D.; Kobayashi, A.; Kato, M. Aromatic versus Aliphatic α-Diimine Ligands in Heteroleptic Copper(I) Emitters: Photophysical and Electrochemical Properties. Anal. Sci. 2020, 36, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Fresta, E.; Weber, M.D.; Fernandez-Cestau, J.; Costa, R.D. White Light-Emitting Electrochemical Cells Based on Deep-Red Cu(I) Complexes. Adv. Opt. Mater. 2019, 7, 1900830. [Google Scholar] [CrossRef]
- Solovyev, I.V.; Kondinski, A.; Monakhov, K.Y.; Koshevoy, I.O.; Grachova, E.V. Synthesis, photophysical properties and cation-binding studies of bipyridine-functionalized gold(i) complexes. Inorg. Chem. Front. 2018, 5, 160–171. [Google Scholar] [CrossRef]
- Belyaev, A.; Slavova, S.O.; Solovyev, I.V.; Sizov, V.V.; Jänis, J.; Grachova, E.V.; Koshevoy, I.O. Solvatochromic dual luminescence of Eu–Au dyads decorated with chromophore phosphines. Inorg. Chem. Front. 2020, 7, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Knall, A.-C.; Kovačič, S.; Hollauf, M.; Reishofer, D.; Saf, R.; Slugovc, C. Inverse electron demand Diels–Alder (iEDDA) functionalisation of macroporous poly(dicyclopentadiene) foams. Chem. Commun. 2013, 49, 7325–7327. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, A.M.; Esquivel, D.; Ouwehand, J.; Van Der Voort, P.; Romero-Salguero, F.J.; Van Deun, R. Temperature dependent NIR emitting lanthanide-PMO/silica hybrid materials. Dalt. Trans. 2017, 46, 7878–7887. [Google Scholar] [CrossRef] [Green Version]
- Khistiaeva, V.V.; Melnikov, A.S.; Slavova, S.O.; Sizov, V.V.; Starova, G.L.; Koshevoy, I.O.; Grachova, E.V. Heteroleptic β-diketonate Ln(iii) complexes decorated with pyridyl substituted pyridazine ligands: Synthesis, structure and luminescence properties. Inorg. Chem. Front. 2018, 5, 3015–3027. [Google Scholar] [CrossRef]
- Evariste, S.; Khalil, A.M.; Kerneis, S.; Xu, C.; Calvez, G.; Costuas, K.; Lescop, C. Luminescent vapochromic single crystal to single crystal transition in one-dimensional coordination polymer featuring the first Cu(i) dimer bridged by an aqua ligand. Inorg. Chem. Front. 2020, 7, 3402–3411. [Google Scholar] [CrossRef]
- Xu, H.B.; Zhang, L.Y.; Ni, J.; Chao, H.Y.; Chen, Z.N. Conformation changes and luminescent properties of Au-Ln (Ln = Nd, Eu, Er, Yb) arrays with 5-ethynyl-2,2′-bipyridine. Inorg. Chem. 2008, 47, 10744–10752. [Google Scholar] [CrossRef]
- Ferrer, M.; Giménez, L.; Gutiérrez, A.; Lima, J.C.; Martínez, M.; Rodríguez, L.; Martín, A.; Puttreddy, R.; Rissanen, K. Polypyridyl-functionalizated alkynyl gold(i) metallaligands supported by tri- and tetradentate phosphanes. Dalt. Trans. 2017, 46, 13920–13934. [Google Scholar] [CrossRef] [Green Version]
- Yersin, H.; Czerwieniec, R.; Shafikov, M.Z.; Suleymanova, A.F. TADF Material Design: Photophysical Background and Case Studies Focusing on Cu I and Ag I Complexes. ChemPhysChem 2017, 18, 3508–3535. [Google Scholar] [CrossRef]
- Hasegawa, Y.; Kitagawa, Y.; Nakanishi, T. Effective photosensitized, electrosensitized, and mechanosensitized luminescence of lanthanide complexes. NPG Asia Mater. 2018, 10, 52–70. [Google Scholar] [CrossRef] [Green Version]
- Nijegorodov, N.; Ramachandran, V.; Winkoun, D.P. The dependence of the absorption and fluorescence parameters, the intersystem crossing and internal conversion rate constants on the number of rings in polyacene molecules. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1997, 53, 1813–1824. [Google Scholar] [CrossRef]
- Márquez, F.; Zicovich-Wilson, C.M.; Corma, A.; Palomares, E.; García, H. Naphthalene Included within All-Silica Zeolites: Influence of the Host on the Naphthalene Photophysics. J. Phys. Chem. B 2001, 105, 9973–9979. [Google Scholar] [CrossRef]
- Pan, Y.Y.; Huang, J.; Wang, Z.M.; Yu, D.W.; Yang, B.; Ma, Y.G. Computational investigation on the large energy gap between the triplet excited-states in acenes. RSC Adv. 2017, 7, 26697–26703. [Google Scholar] [CrossRef] [Green Version]
- Yuasa, J.; Dan, M.; Kawai, T. Phosphorescent properties of metal-free diphosphine ligands and effects of copper binding. Dalt. Trans. 2013, 42, 16096. [Google Scholar] [CrossRef]
- Kozhevnikov, V.N.; Shabunina, O.V.; Kopchuk, D.S.; Ustinova, M.M.; König, B.; Kozhevnikov, D.N. Facile synthesis of 6-aryl-3-pyridyl-1,2,4-triazines as a key step toward highly fluorescent 5-substituted bipyridines and their Zn(II) and Ru(II) complexes. Tetrahedron 2008, 64, 8963–8973. [Google Scholar] [CrossRef]
- CrysAlisPro, Rigaku Oxford Diffraction; Version 1.171.39.35a; Agilent Technologies: Santa Clara, CA, USA, 2017.
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Sillen, A.; Engelborghs, Y. The Correct Use of “Average” Fluorescence Parameters. Photochem. Photobiol. 1998, 67, 475–486. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Andrae, D.; Haussermann, U.; Dolg, M.; Stoll, H.; Preuss, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Mennucci, B.; Cancès, E.; Tomasi, J. Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar] [CrossRef]
- Cancès, E.; Mennucci, B.; Tomasi, J. A new integral equation formalism for the polarizable continuum model: Theoretical background and applications to isotropic and anisotropic dielectrics. J. Chem. Phys. 1997, 107, 3032–3041. [Google Scholar] [CrossRef]
- Martin, R.L.R.L. Natural transition orbitals. J. Chem. Phys. 2003, 118, 4775–4777. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | ||
|---|---|---|
| 1 | 0.83 | shoulder at ca. 1.2 |
| 2 | 0.81 | 1.31 |
| 3 | 0.87 | 1.1; 1.5 |
| 4 | 0.86 | 1.29 |
| 5 | 0.72 | 1.41 |
| 6 | 0.80 | - |
| 295 K | 77 K | ||||
|---|---|---|---|---|---|
| λem, nm | Φ, % | τav, µs | λem, nm | τav, µs | |
| 1 | 580 | 2.6 | 4.2 | 602 | 26.5 |
| 2 | 622 | 0.6 | 1.1 | 640 | 20.1 |
| 3 | 578 | 0.2 | 2.6 | 516 * | 1096.4 |
| 4 | 510 ‡; 600 | 0.6 | 3.8 | 420; 547 ‡; 600 | 268.5 |
| 5 | 445,§ 600 | 0.5 | 1.1 | 445,# 613 | 130.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paderina, A.; Melnikov, A.; Slavova, S.; Sizov, V.; Gurzhiy, V.; Petrovskii, S.; Luginin, M.; Levin, O.; Koshevoy, I.; Grachova, E. The Tail Wags the Dog: The Far Periphery of the Coordination Environment Manipulates the Photophysical Properties of Heteroleptic Cu(I) Complexes. Molecules 2022, 27, 2250. https://doi.org/10.3390/molecules27072250
Paderina A, Melnikov A, Slavova S, Sizov V, Gurzhiy V, Petrovskii S, Luginin M, Levin O, Koshevoy I, Grachova E. The Tail Wags the Dog: The Far Periphery of the Coordination Environment Manipulates the Photophysical Properties of Heteroleptic Cu(I) Complexes. Molecules. 2022; 27(7):2250. https://doi.org/10.3390/molecules27072250
Chicago/Turabian StylePaderina, Aleksandra, Alexey Melnikov, Sofia Slavova, Vladimir Sizov, Vladislav Gurzhiy, Stanislav Petrovskii, Maksim Luginin, Oleg Levin, Igor Koshevoy, and Elena Grachova. 2022. "The Tail Wags the Dog: The Far Periphery of the Coordination Environment Manipulates the Photophysical Properties of Heteroleptic Cu(I) Complexes" Molecules 27, no. 7: 2250. https://doi.org/10.3390/molecules27072250