
Coumarin-Palladium(II) Complex Acts as a Potent and Non-Toxic Anticancer Agent against Pancreatic Carcinoma Cells

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Coumarin-Palladium(II) Complex Potently Reduces Viability and Proliferation of Pancreatic Carcinoma Cells In Vitro
2.2. Coumarin-Palladium(II) Complex Impairs Migratory Potential of Pancreatic Carcinoma Cells In Vitro
2.3. Apoptosis Is Induced upon the Treatment of PANC-1 with Coumarin-Palladium(II) Complex
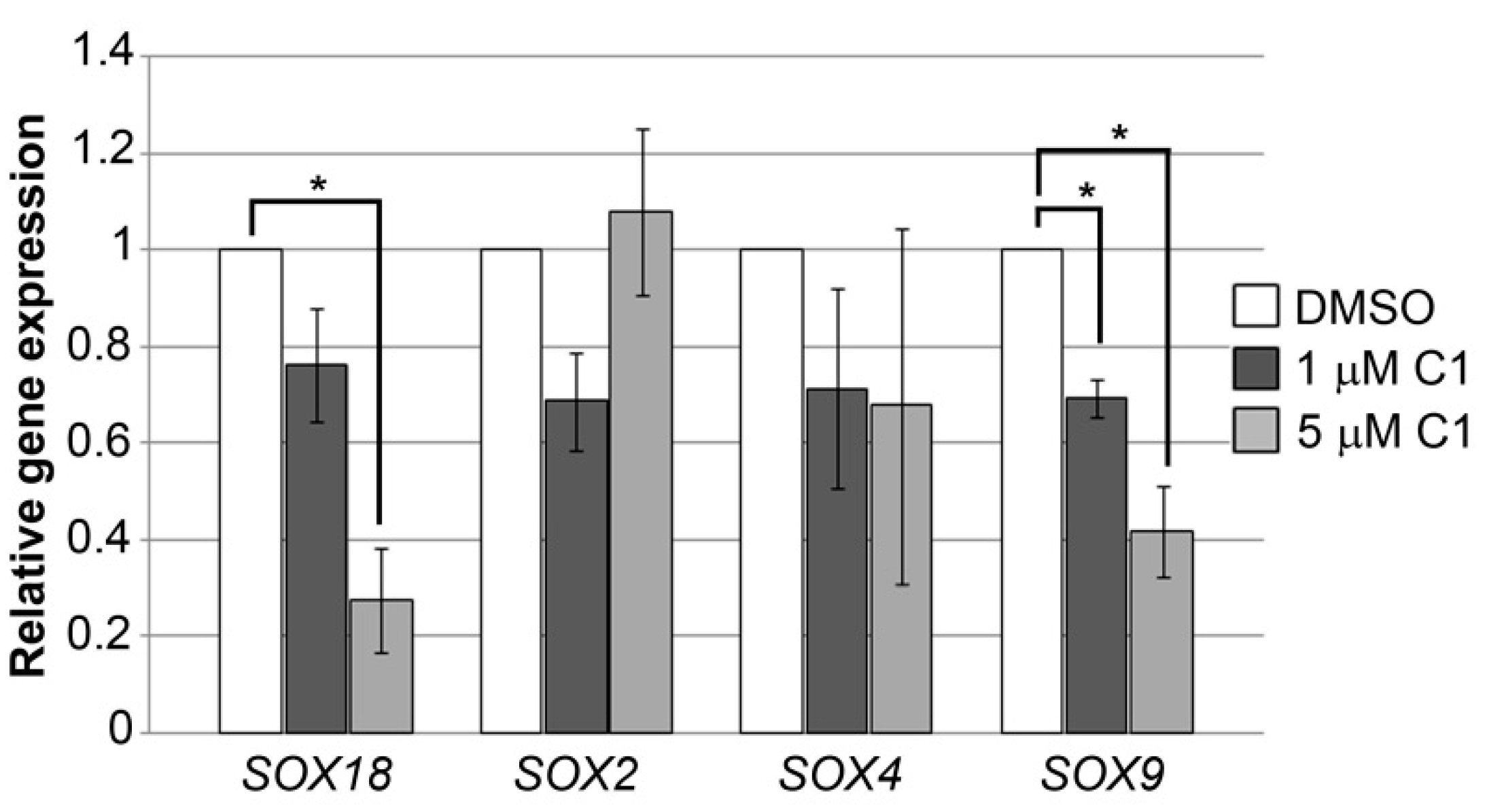
2.4. Coumarin-Palladium(II) Complex Significantly Reduces SOX18 and SOX9 Expression in Pancreatic Carcinoma Cells In Vitro
2.5. Anticancer Activity of Coumarin-Palladium(II) Complex in the Zebrafish Xenograft Model of Pancreatic Carcinoma
2.6. Hepatotoxicity Assessment
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Colony Formation Assay
4.5. Wound-Healing Assay
4.6. Apoptosis Assay
4.7. qRT-PCR
4.8. Zebrafish Maintenance
4.9. Anticancer Activity Evaluation in PANC-1-Zebrafish Xenografts
4.10. In Vivo Hepatotoxicity Test
4.11. Statistical Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ilic, M.; Ilic, I. Epidemiology of pancreatic cancer. World J. Gastroenterol. 2016, 22, 9694–9705. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Bauer, C.A.; Öhlund, D.; Lauth, M.; Buchholz, M.; Michl, P.; Tuveson, D.A.; Gress, T. Stromal biology and therapy in pancreatic cancer: Ready for clinical translation? Gut 2018, 68, 159–171. [Google Scholar] [CrossRef]
- Torphy, R.J.; Fujiwara, Y.; Schulick, R.D. Pancreatic cancer treatment: Better, but a long way to go. Surg. Today 2020, 50, 1117–1125. [Google Scholar] [CrossRef]
- Riveiro, M.; De Kimpe, N.; Moglioni, A.; Vazquez, R.; Monczor, F.; Shayo, C.; Davio, C. Coumarins: Old compounds with novel promising therapeutic perspectives. Curr. Med. Chem. 2010, 17, 1325–1338. [Google Scholar] [CrossRef]
- Banikazemi, Z.; Mirazimi, S.M.; Dashti, F.; Mazandaranian, M.R.; Akbari, M.; Morshedi, K.; Aslanbeigi, F.; Rashidian, A.; Chamanara, M.; Hamblin, M.R.; et al. Coumarins and Gastrointestinal Cancer: A New Therapeutic Option? Front. Oncol. 2021, 11, 752784. [Google Scholar] [CrossRef]
- Devji, T.; Reddy, C.; Woo, C.; Awale, S.; Kadota, S.; Carrico-Moniz, D. Pancreatic anticancer activity of a novel geranylgeranylated coumarin derivative. Bioorg. Med. Chem. Lett. 2011, 21, 5770–5773. [Google Scholar] [CrossRef]
- Jun, M.; Bacay, A.F.; Moyer, J.; Webb, A.; Carrico-Moniz, D. Synthesis and biological evaluation of isoprenylated coumarins as potential anti-pancreatic cancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 4654–4658. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, R.; Jun, M.; Bacay, A.F.; Eyring, K.; Webb, A.; Carrico-Moniz, D. Identification of the Factors Responsible for the Selective in vitro Cytotoxic Activity of Isoprenylated Coumarin Derivatives under Nutrient-deprived Conditions. J. Cancer 2016, 7, 160–166. [Google Scholar] [CrossRef] [Green Version]
- Balewski, Ł.; Szulta, S.; Jalińska, A.; Kornicka, A. A Mini-Review: Recent Advances in Coumarin-Metal Complexes With Biological Properties. Front. Chem. 2021, 9, 781779. [Google Scholar] [CrossRef]
- Avdović, E.H.; Petrović, I.P.; Stevanović, M.J.; Saso, L.; Dimitric Marković, J.M.; Filipović, N.D.; Živić, M.; Antić, T.N.C.; Žižić, M.V.; Todorović, N.V.; et al. Synthesis and Biological Screening of New 4-Hydroxycoumarin Derivatives and Their Palladium(II) Complexes. Oxidative Med. Cell. Longev. 2021, 2021, 8849568. [Google Scholar] [CrossRef] [PubMed]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, C.; Cordani, M.; Padroni, C.; Blandino, G.; Di Agostino, S.; Donadelli, M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim. Biophys. Acta Mol. Cell Res. 2015, 1853, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Edelman, H.E.; McClymont, S.A.; Tucker, T.R.; Pineda, S.; Beer, R.L.; McCallion, A.S.; Parsons, M.J. SOX9 modulates cancer biomarker and cilia genes in pancreatic cancer. Hum. Mol. Genet. 2021, 30, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Cheng, Y.-Y.; Liao, W.-C.; Tien, Y.-W.; Yang, C.-H.J.; Hsu, S.-M.; Huang, P.-H. SOX4 Transcriptionally regulates multiple SEMA3/plexin family members and promotes tumor growth in pancreatic cancer. PLoS ONE 2012, 7, e48637. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Gu, D.; Wan, J.; Yu, B.; Zhang, X.; Chiorean, E.G.; Wang, Y.; Xie, J. The role of GLI-SOX2 signaling axis for gemcitabine resistance in pancreatic cancer. Oncogene 2019, 38, 1764–1777. [Google Scholar] [CrossRef] [Green Version]
- McDonald, E.; Krishnamurthy, M.; Goodyer, C.G.; Wang, R. The Emerging role of SOX transcription factors in pancreatic endocrine cell development and function. Stem Cells Dev. 2009, 18, 1379–1388. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, H.; Zhang, D.; Yu, X.; Leng, X.; Li, S.; Zhu, W. Overexpression of SOX18 correlates with accelerated cell growth and poor prognosis in human pancreatic ductal adenocarcinoma. Biochem. Biophys. Res. Commun. 2016, 479, 510–516. [Google Scholar] [CrossRef]
- White, R.; Rose, K.; Zon, L. Zebrafish cancer: The state of the art and the path forward. Nat. Rev. Cancer 2013, 13, 624–636. [Google Scholar] [CrossRef]
- Binenbaum, Y.; Na’ara, S.; Gil, Z. Gemcitabine resistance in pancreatic ductal adenocarcinoma. Drug Resist. Updat. 2015, 23, 55–68. [Google Scholar] [CrossRef]
- Aung, K.L.; Fischer, S.E.; Denroche, R.E.; Jang, G.-H.; Dodd, A.; Creighton, S.; Southwood, B.; Liang, S.-B.; Chadwick, D.; Zhang, A.; et al. Genomics-Driven Precision Medicine for Advanced Pancreatic Cancer: Early Results from the COMPASS Trial. Clin. Cancer Res. 2018, 24, 1344–1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.-L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Ough, M.; Li, L.; Hinkhouse, M.M.; Ritchie, J.M.; Spitz, D.; Cullen, J.J. Treatment of pancreatic cancer cells with dicumarol induces cytotoxicity and oxidative stress. Clin. Cancer Res. 2004, 10, 4550–4558. [Google Scholar] [CrossRef] [Green Version]
- Awale, S.; Okada, T.; Dibwe, D.F.; Maruyama, T.; Takahara, S.; Okada, T.; Endo, S.; Toyooka, N. Design and synthesis of functionalized coumarins as potential anti-austerity agents that eliminates cancer cells’ tolerance to nutrition starvation. Bioorg. Med. Chem. Lett. 2019, 29, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Ulukaya, E.; Ari, F.; Dimas, K.; Ikitimur, E.I.; Guney, E.; Yilmaz, V.T. Anti-cancer activity of a novel palladium(II) complex on human breast cancer cells in vitro and in vivo. Eur. J. Med. Chem. 2011, 46, 4957–4963. [Google Scholar] [CrossRef]
- Sahin, Ö.; Özdemir, Ü.Ö.; Seferoğlu, N.; Genc, Z.K.; Kaya, K.; Aydiner, B.; Tekin, S.; Seferoğlu, Z. New platinum (II) and palladium (II) complexes of coumarin-thiazole Schiff base with a fluorescent chemosensor properties: Synthesis, spectroscopic characterization, X-ray structure determination, in vitro anticancer activity on various human carcinoma cell lines and computational studies. J. Photochem. Photobiol. B Biol. 2018, 178, 428–439. [Google Scholar] [CrossRef]
- Gramni, L.; Vukea, N.; Chakraborty, A.; Samson, W.; Dingle, L.M.K.; Xulu, B.; de la Mare, J.-A.; Edkins, A.L.; Booysen, I.N. Anticancer evaluation of ruthenium(III) complexes with N-donor ligands tethered to coumarin or uracil moieties. Inorg. Chim. Acta 2019, 492, 98–107. [Google Scholar] [CrossRef]
- Lu, W.; Shi, J.; Nie, Y.; Yang, L.; Chen, J.; Zhao, F.; Yang, S.; Xu, L.; Chi, X. Synthesis, crystal structure, antiproliferative activity, DNA binding and density functional theory calculations of 3-(pyridin-2-yl)-8-tert-butylcoumarin and its copper(II) complex. Appl. Organomet. Chem. 2020, 34, e5875. [Google Scholar] [CrossRef]
- Mestizo, P.D.; Narváez, D.M.; Pinzón-Ulloa, J.A.; Di Bello, D.T.; Franco-Ulloa, S.; Macías, M.A.; Groot, H.; Miscione, G.P.; Suescun, L.; Hurtado, J.J. Novel complexes with ONNO tetradentate coumarin schiff-base donor ligands: X-ray structures, DFT calculations, molecular dynamics and potential anticarcinogenic activity. BioMetals 2021, 34, 119–140. [Google Scholar] [CrossRef]
- Bredholt, G.; Mannelqvist, M.; Stefansson, I.M.; Birkeland, E.; Bø, T.H.; Oyan, A.M.; Trovik, J.; Kalland, K.-H.; Jonassen, I.; Salvesen, H.B.; et al. Tumor necrosis is an important hallmark of aggressive endometrial cancer and associates with hypoxia, angiogenesis and inflammation responses. Oncotarget 2015, 6, 39676–39691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsch-Bluman, A.; Feiglin, A.; Arbib, E.; Stern, T.; Shoval, H.; Schwob, O.; Berger, M.; Benny, O. Tissue necrosis and its role in cancer progression. Oncogene 2019, 38, 1920–1935. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Hiramoto, A.; Kim, H.-S.; Wataya, Y. Anticancer Strategy Targeting Cell Death Regulators: Switching the Mechanism of Anticancer Floxuridine-Induced Cell Death from Necrosis to Apoptosis. Int. J. Mol. Sci. 2020, 21, 5876. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A Link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Akkol, E.K.; Genç, Y.; Karpuz, B.; Sobarzo-Sánchez, E.; Capasso, R. Coumarins and coumarin-related compounds in pharmacotherapy of cancer. Cancers 2020, 12, 1959. [Google Scholar] [CrossRef]
- Del Poeta, G.; Venditti, A.; Del Principe, M.I.; Maurillo, L.; Buccisano, F.; Tamburini, A.; Cox, M.C.; Franchi, A.; Bruno, A.; Mazzone, C.; et al. Amount of spontaneous apoptosis detected by Bax/Bcl-2 ratio predicts outcome in acute myeloid leukemia (AML). Blood 2003, 101, 2125–2131. [Google Scholar] [CrossRef]
- Diaz, J.-L.; Oltersdorf, T.; Horne, W.; McConnell, M.; Wilson, G.; Weeks, S.; Garcia, T.; Fritz, L.C. A Common Binding Site Mediates Heterodimerization and Homodimerization of Bcl-2 Family Members. J. Biol. Chem. 1997, 272, 11350–11355. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2017, 25, 27–36. [Google Scholar] [CrossRef] [Green Version]
- De Jong, Y.; Monderer, D.; Brandinelli, E.; Monchanin, M.; van den Akker, B.E.; van Oosterwijk, J.; Blay, J.Y.; Dutour, A.; Bovée, J.V.M.G. Bcl-xl as the most promising Bcl-2 family member in targeted treatment of chondrosarcoma. Oncogenesis 2018, 7, 74. [Google Scholar] [CrossRef]
- de Carné Trécesson, S.; Souazé, F.; Basseville, A.; Bernard, A.-C.; Pécot, J.; Lopez, J.; Bessou, M.; Sarosiek, K.; Letai, A.; Barille-Nion, S.; et al. BCL-XL directly modulates RAS signalling to favour cancer cell stemness. Nat. Commun. 2017, 8, 1123. [Google Scholar] [CrossRef]
- Findley, H.W.; Gu, L.; Yeager, A.M.; Zhou, M. Expression and regulation of Bcl-2, Bcl-xl, and Bax correlate with p53 status and sensitivity to apoptosis in childhood acute lymphoblastic leukemia. Blood 1997, 89, 2986–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyashita, T.; Harigai, M.; Hanada, M.; Reed, J.C. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res. 1994, 54, 3131–3135. [Google Scholar]
- Lee, J.U.; Hosotani, R.; Wada, M.; Doi, R.; Kosiba, T.; Fujimoto, K.; Miyamoto, Y.; Mori, C.; Nakamura, N.; Shiota, K.; et al. Mechanism of apoptosis induced by cisplatin and VP-16 in PANC-1 cells. Anticancer Res. 1997, 17, 3445–3450. [Google Scholar] [PubMed]
- Lee, J.-U.; Hosotani, R.; Wada, M.; Doi, R.; Kosiba, T.; Fujimoto, K.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Nishimura, Y.; et al. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur. J. Cancer 1999, 35, 1374–1380. [Google Scholar] [CrossRef]
- Feldmann, G.; Beaty, R.; Hruban, R.H.; Maitra, A. Molecular genetics of pancreatic intraepithelial neoplasia. J. Hepato-Biliary-Pancreat. Surg. 2007, 14, 224–232. [Google Scholar] [CrossRef] [Green Version]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Qin, Y.; Ji, S.; Ling, J.; Fu, J.; Zhuang, Z.; Fan, X.; Song, L.; Yu, X.; Chiao, P.J. SOX9 activity is induced by oncogenic Kras to affect MDC1 and MCMs expression in pancreatic cancer. Oncogene 2018, 37, 912–923. [Google Scholar] [CrossRef]
- Seymour, P.A. Sox9: A master regulator of the pancreatic program. Rev. Diabet. Stud. 2014, 11, 51–83. [Google Scholar] [CrossRef] [Green Version]
- Kaushik, G.; Seshacharyulu, P.; Rauth, S.; Nallasamy, P.; Rachagani, S.; Nimmakayala, R.K.; Vengoji, R.; Mallya, K.; Chirravuri-Venkata, R.; Singh, A.B.; et al. Selective inhibition of stemness through EGFR/FOXA2/SOX9 axis reduces pancreatic cancer metastasis. Oncogene 2021, 40, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Sukdolak, S.; Solujić, S.; Manojlović, N.; Vuković, N.; Krstić, L.J. Hantzsch reaction of 3-(2-bromoacetyl)-4-hydroxy-chromen-2-one. Synthesis of 3-(thiazol-4-yl)-4-hydroxy coumarines. J. Heterocycl. Chem. 2004, 41, 593–596. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, L.; He, Q.; Chen, W.; Sun, C.; Wang, X.; Chen, X.; Wang, R.; Hsiao, C.-D.; Liu, K. A rapid assessment for predicting drug-induced hepatotoxicity using zebrafish. J. Pharmacol. Toxicol. Methods 2017, 84, 102–110. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GAPDH F | 5′-GCC TCA AGA TCA TCA GCA ATG C-3′ |
| GAPDH R | 5′-CCA CGA TAC CAA AGT TGT CAT GG-3′ |
| SOX9 F | 5’-CTT CTG AAC GAG AGC GAG A-3’ |
| SOX9 R | 5’-CTG CCC GTT CTT CAC CGA CTT C-3’ |
| SOX4 F | 5′-CCA AAT CTT TTG GGG ACT TTT-3′ |
| SOX4 R | 5’-CTG GCC CCT CAA CTC CTC-3′ |
| qSOX2 F | 5′-CCC CTG GCA TGG CTC TTG GC-3′ |
| qSOX2 R | 5′-TCG GCG CCG GGG AGA TAC AT-3′ |
| SOX18 F | 5′-TTC CAT GTC ACA GCC CCC TAG-3′ |
| SOX18 R | 5′-GAC ACG TGG GAA CTC CAG-3′ |
| BAX F | 5′-TGG CAG CTG ACA TGT TTT CTG AC-3′ |
| BAX R | 5′-TCA CCC AAC CAC CCT GGT CTT-3′ |
| BCL-2 F | 5′-TCG CCC TGT GGA TGA CTG A-3′ |
| BCL-2 R | 5′-CAG AGA CAG CCA GGA GAA ATC-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krstic, A.; Pavic, A.; Avdovic, E.; Markovic, Z.; Stevanovic, M.; Petrovic, I. Coumarin-Palladium(II) Complex Acts as a Potent and Non-Toxic Anticancer Agent against Pancreatic Carcinoma Cells. Molecules 2022, 27, 2115. https://doi.org/10.3390/molecules27072115
Krstic A, Pavic A, Avdovic E, Markovic Z, Stevanovic M, Petrovic I. Coumarin-Palladium(II) Complex Acts as a Potent and Non-Toxic Anticancer Agent against Pancreatic Carcinoma Cells. Molecules. 2022; 27(7):2115. https://doi.org/10.3390/molecules27072115
Chicago/Turabian StyleKrstic, Aleksandra, Aleksandar Pavic, Edina Avdovic, Zoran Markovic, Milena Stevanovic, and Isidora Petrovic. 2022. "Coumarin-Palladium(II) Complex Acts as a Potent and Non-Toxic Anticancer Agent against Pancreatic Carcinoma Cells" Molecules 27, no. 7: 2115. https://doi.org/10.3390/molecules27072115