3.2. Synthesis
3.2.1. Synthesis of tert-Butyl Substituted Nitrones (General Method)
A methyl-4-nitroheptanoate (1a) or dimethyl 4-nitroheptanedioate (1b) (100 mmol) was added dropwise to freshly prepared solution of sodium methylate in methanol (0.35 M, 40 mL) within 15 min. The yellow solution formed was stirred for 20 min at room temperature, then a solution of 4,4-dimethylpent-1-en-3-one 2 (100 mmol) in 40 mL of dry methanol was added dropwise within 30 min. The reaction mixture was allowed to cool down to room temperature, then heated to reflux for 3 h. (TLC control on SiO2, chloroform, stained with phosphomolybdic acid). The pH was adjusted to neutral by addition of glacial acetic acid (ca. 1.5 mL). and the solvent was distilled off in vacuum. The residue was dissolved in ethyl acetate (150 mL) and successively washed with water (70 mL), saturated solution of sodium bicarbonate (100 mL) and again with water (100 mL). The organic layer was dried with anhydrous Na2SO4 and the solvent was evaporated in vacuum. The residue was dissolved in 30 mL of THF and mixed with aqueous solution ammonium chloride (1.7 M, 60 mL). The resulting solution was cooled to 7 °C, and zinc dust (24.0 g, 370 mmol) was added by small portion maintaining the temperature in the range of 7–15 °C. The reaction mixture was stirred for 1 h at 5 °C and then 1 h at room temperature. Inorganic precipitate was filtered off and washed with ethanol (200 mL). The filtrate was concentrated in vacuum and the residue was dissolved in water (50 mL). Water solution was extracted with diethyl ether (50 mL) and organic layer was discarded. Water layer was saturated by NaCl and extracted with CHCl3 (3 × 100 mL). The combined organic extracts were dried with anhydrous Na2SO4. The solvent was evaporated in vacuum, the crude residue was purified by column chromatography (SiO2, chloroform-methanol 40:1 mixture as an eluent, detected under UV lamp) to give 4a or 4b.
Methyl 3-(5-tert-butyl-2-ethyl-1-oxido-3,4-dihydro-2H-pyrrol-2-yl)propanoate (4a): yield 15.3 g (60%). Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C14H25NO3)+: 255.1829; found: 255.1831. IR (neat) νmax: 1737 (C=O), 1573 (C=N). 1H NMR (500 MHz; CDCl3, δ): 0.77 (t, Jt = 7.4 Hz, 3H), 1.22 (s, 9H, t-Bu), 1.41–1.50 (m, 1H), 1.73–1.82 (m, 2H), 1.83–1.92 (m, 2H), 2.07–2.15 (m, 1H), 2.16–2.28 (m, 2H), 2.51 (dd, Jd1 = 7.6 Hz, Jd2 = 7.2 Hz, 2H), 3.58 (s, 3H, OCH3). 13CNMR (125 MHz; CDCl3, δ): 7.29, 23.98, 25.26, 27.31, 28.23, 30.50, 32.67, 33.33, 51.38, 79.25, 150.67, 173.3.
Dimethyl 3,3′-(5-tert-butyl-1-oxido-3,4-dihydro-2H-pyrrole-2,2-diyl)dipropanoate (4b): yield 17.8 g (57%). Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C16H27NO5)+: 313.1884; found: 313.1880. IR (neat) νmax: 1737 (C=O), 1573 (C=N). 1H NMR (400 MHz; CDCl3, δ): 1.15 (s, 9H, t-Bu), 1.71–1.82 (m, 4H), 2.02–2.11 (m, 2H), 2.11–2.19 (m, 4H), 2.45–2.51 (m, 2H), 3.52 (s, 6H, OCH3). 13CNMR (100 MHz; CDCl3, δ): 24.44, 25.15, 27.13, 28.07, 32.53, 33.35, 51.49, 78.04, 151.06, 173.02.
3.2.2. 3-(5-tert-Butyl-2-ethyl-1-oxido-3,4-dihydro-2H-pyrrol-2-yl)propanoic Acid (5a)
An aqueous solution of sodium hydroxide (3.2 M, 50 mL) was added to the solution of 4a (10.2 g, 40 mmol) in methanol (50 mL), and the resulting homogeneous mixture was left at room temperature for 24 h. Methanol was distilled off in vacuum, the water solution was shaken with diethyl ether (50 mL), and the organic layer was discarded. Chloroform (70 mL) was added to the water layer and 2 M aqueous solution of H2SO4(40 mL) was added to the mixture upon stirring. The organic layer was separated, and the water layer was extracted with CHCl3 (2 × 50 mL). Combined organic extracts were dried with anhydrous Na2SO4, the solvent was evaporated in vacuum, the crude residue was treated with diethyl ether (30 mL), and and white crystalline precipitate of 5a was filtered off, yield 8.5 g, (91%), m.p. 160.6–162.0 °C (ethyl acetate). Found: C, 64.91; H, 9.31; N, 5.93; calcd. For C13H23NO3: C, 64.70; H, 9.61; N, 5.80%; IR (KBr) νmax: 1726 (C=O), 1594 (C=N); 1H NMR (500 MHz; CDCl3, δ): 0.79 (t, Jt = 7.4 Hz, 3H), 1.26 (s, 9H, t-Bu), 1.46–1.55 (m, 1H), 1.76–1.86 (m, 2H), 1.87–1.96 (m, 2H), 2.09–2.21 (m, 2H), 2.26–2.34 (m, 1H), 2.54–2.66 (m, 2H), 11.16 (br. S, 1H). 13CNMR (125 MHz; CDCl3, δ): 7.30, 24.01, 25.55, 28.03, 28.76, 30.55, 32.10, 33.84, 80.39, 157.62, 174.74.
3.2.3. 3,3′-(5-tert-Butyl-1-oxido-3,4-dihydro-2H-pyrrole-2,2-diyl)dipropanoic Acid (5b)
An aqueous solution of sodium hydroxide (3.2 M, 5 mL) was added to the solution of 4b (1.25 g, 4 mmol) in methanol (5 mL). The resulting homogeneous solution was allowed to stand at room temperature for 24 h, methanol was distilled off in vacuum pressure and 2M solution of H2SO4 (4.5 mL) was added to the residue upon stirring. The formed precipitate of 5b was filtered off and successively washed with ice-cold water (2 mL) and diethyl ether (10 mL) and dried in vacuum, yield 1.0 g (95%). White crystalline solid, m.p. 223.3–224.5 °C (ethanol). Found: C, 59.22; H, 8.29; N, 4.78; calcd. For C14H23NO5: C, 58.93; H, 8.12; N, 4.91%; IR (KBr) νmax: 1720 (C=O), 1594 (C=N). 1H NMR (300 MHz; (CD3)2SO, δ): 1.20 (s, 9H, t-Bu), 1.62–1.76 (m, 2H), 1.80–2.05 (m, 6H), 2.16–2.32 (m, 2H), 2.54–2.67 (m, 2H). 13CNMR (75 MHz; (CD3)2SO, δ): 23.66, 24.89, 26.15, 26.78, 28.00, 32.71, 33.07, 77.91, 150.65, 174.17
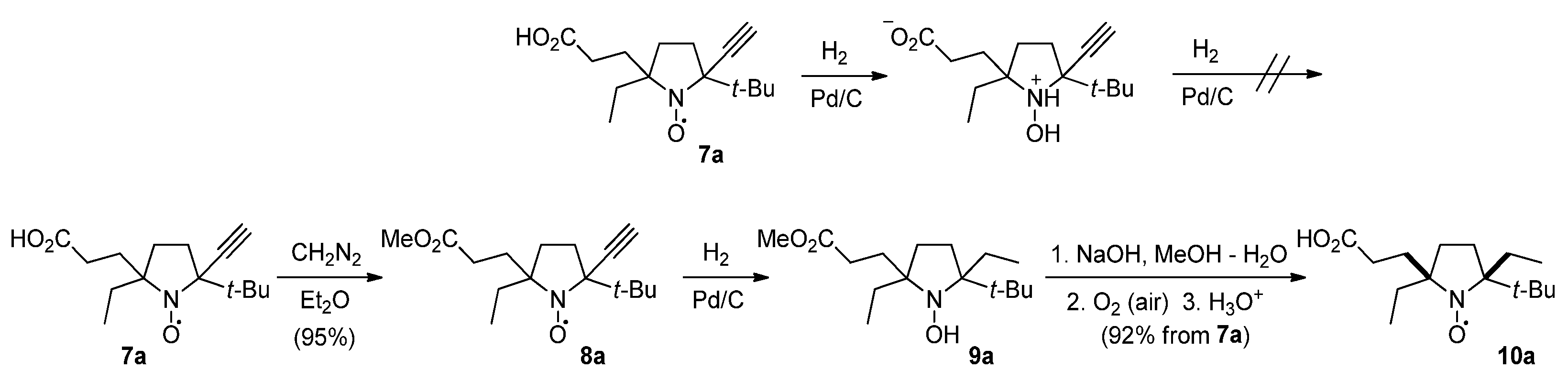
3.2.4. 2-tert-Butyl-5-(2-carboxyethyl)-5-ethyl-2-ethynylpyrrolidine-1-oxyl (7a,a′)
A powder of 5a (3.6 g, 15 mmol) was added to a 0.5–1 M solution of ethynyl-magnesium bromide in THF (250 mL) upon stirring. The mixture was allowed to stand at room temperature for 168 h (TLC control on SiO2, ethyl acetate: methanol: acetic acid 100:10:1, detected under UV lamp), then quenched with water (20 mL) and acidified with saturated aqueous sodium bisulfate solution (150 mL) to pH 3–4. The organic layer was separated, and the water phase was extracted with ethyl acetate (2 × 100 mL). The combined organic layers were dried with anhydrous Na2SO4. The solvent was evaporated in vacuum and the crude residue was dissolved in methanol (70 mL) and basified with sodium hydroxide solution (1 M, 20 mL). Methylene blue (6 mg, 0.02 mmol) was added to the mixture and the air was bubbled until the solution turned dark blue. The methanol was distilled off in vacuum, and the remaining aqueous solution was washed with diethyl ether (30 mL), acidified with saturated aqueous sodium bisulfate solution (20 mL) to pH < 4, and extracted with ethyl acetate (3 × 50 mL). The organic phase was dried with Na2SO4 and the solvent was evaporated in vacuum. The residue was purified by column chromatography (SiO2, eluent ethyl acetate: methanol: acetic acid 100:10:1) to give of 7a,a′, yield 2.40 g (60%), yellow crystalline solid, m.p. 109.9–112.3 °C (hexane: ethyl acetate 5:1). Found: C, 67.66; H, 9.04; N, 5.22; calcd. for C15H24NO3: C, 67.64; H, 9.08; N, 5.26%; IR (KBr) νmax: 3309, 2983, 2969, 2956, 1718, 1214, 619. 1H NMR(400 MHz; CD3OD/CDCl3, Zn/CF3COOH system δ): 0.63 (t, Jt = 7.5 Hz, 3H), 0.86 (s, 9H, t-Bu), 1.34–1.53 (m, 1H), 1.53–1.67 (m, 2H), 1.71–1.85 (m, 3H), 1.89–2.06 (m, 1H), 2.06–2.16 (m, 1H), 2.23–2.29 (m, 2H), 2.75 (s, 1H). Signals of minor isomer: 0.50 (t, Jt = 7.5 Hz, 3H), 0.67 (s, 9H, t-Bu), 2.64 (s, 1H).
3.2.5. 2-tert-Butyl-5-ethyl-2-ethynyl-5-(3-methoxy-3-oxopropyl)pyrrolidine-1-oxyl (8a)
A solution of 7a (1.0 g, 3.76 mmol) in diethyl ether (10 mL) was slowly added to the 0.4 M solution (40 mL) of diazomethane in diethyl ether at 0 °C. The reaction mixture was stirred for 1 h at this temperature. The reaction was monitored by TLC (SiO2, ethyl acetate: methanol: acetic acid 200:20:1 mixture; detected under UV lamp). Then, acetic acid (2 mL) was slowly added to remove excess of diazomethane, and the resulting solution was washed with 10% aqueous sodium bicarbonate (2 × 25 mL) and water (1 × 25 mL). The organic phase was dried with Na2SO4, and the solvent was evaporated in vacuum. The residue was purified by column chromatography (SiO2, ethyl acetate as an eluent, detected under UV lamp) to give 8a, yield 1.0 g (95%), yellow crystalline solid, m.p. 65.4–66.2 °C (hexane). Found: C, 68.79; H, 9.22; N, 4.97; calcd. for C16H26NO3: C, 68.54; H, 9.35; N, 5.00%; IR (KBr) νmax: 3259 (≡C-H), 1727 (C=O). 1H NMR (500 MHz; CD3OD/CDCl3, Zn/CF3COOH system δ): 0.97 (t, Jt = 7.5 Hz, 3H), 1.20 (s, 9H, t-Bu), 1.86–2.26 (m, 6H), 2.26–2.47 (m, 2H), 2.60–2.68 (m, 2H), 3.12 (s, 1H), 3.74 (s, 3H).
3.2.6. 2-tert-Butyl-5-(2-carboxyethyl)-2,5-diethylpyrrolidine-1-oxyl (10a)
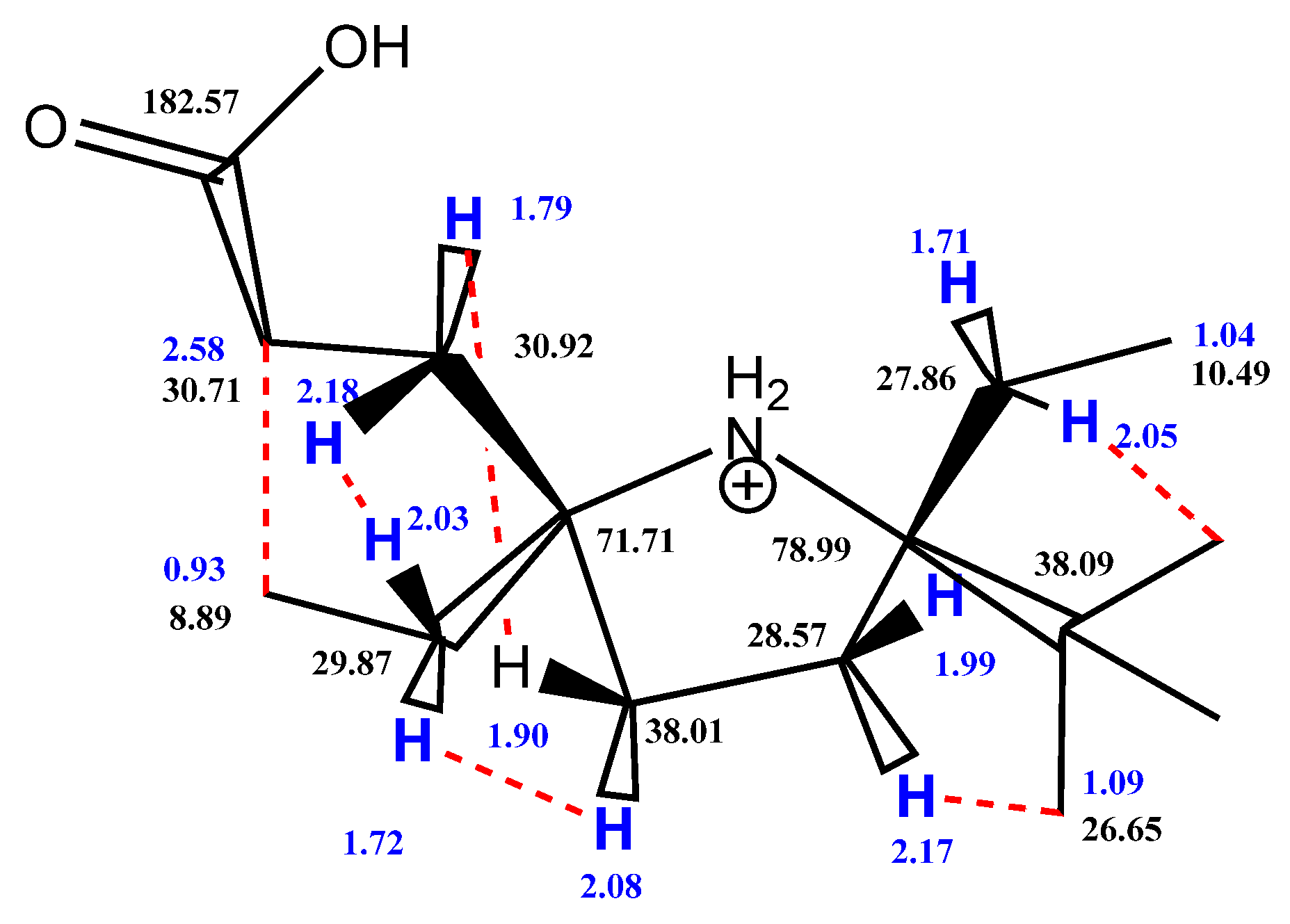
A solution of 8a (1.0 g, 3.6 mmol) in methanol (10 mL) was placed in the reaction vessel equipped with magnetic stirrer and connection line to gasometer filled with hydrogen. The catalyst (Pd/C, 4%, 30 mg) was added, and the system was purged with hydrogen and closed. The mixture was vigorously stirred until hydrogen absorption ceased (ca. 7 h, 0.22 L of hydrogen absorbed), then the catalyst was filtered off and washed with methanol. Filtrate was mixed with 2M aqueous solution of sodium hydroxide (10 mL) and allowed to stand at room temperature for 10 h. The solution turned yellow. Methanol was distilled off in vacuum, the remaining water solution was shaken with diethyl ether (15 mL) and organic layer was discarded. Chloroform (20 mL) was added to the mixture was acidified with saturated aqueous sodium bisulfate solution (10 mL) to pH < 4 upon stirring. Organic layer was separated and water layer was extracted with chloroform (20 mL). Combined extract was dried with Na2SO4 and the solvent was evaporated in vacuum. The residue was purified by column chromatography (SiO2, ethyl acetate: methanol: aceticacid100:10:1 mixture as an eluent) to give 10a, yield 0.90 g (92%), yellow crystalline solid,. m.p. 101–102 °C dec. (hexane). Found: C, 66.81; H, 10.49; N, 5.14; calcd. for C15H28NO3: C, 66.63; H, 10.44; N, 5.18%; IR (KBr) ν max: 1704 (C=O). 1H NMR (400 MHz; CD3OD/CDCl3, Zn/CF3COOH system δ): 0.71 (t, Jt = 7.4 Hz, 3H), 0.84 (t, Jt = 7.5 Hz, 3H), 0.88 (s, 9H, t-Bu), 1.43–1.60 (m, 3H), 1.63–1.71 (m, 1H), 1.74–2.03 (m, 6H), 2.29–2.45 (m, 2H).
3.2.7. Synthesis of tert-Butyl Substituted Nitrones, Containing 3-Hydroxypropyl Moiety (General Method)
A solution of corresponding nitrone 4a,b (10 mmol) in anhydrous diethyl ether (10 mL) was added dropwise within 20 min to a suspension of lithium aluminum hydride (1.14 g, 30 mmol) in anhydrous diethyl ether (30 mL) upon stirring. After the spontaneous boiling of the reaction mixture ceased, it was heated to reflux and stirred for 4 h. The reaction was monitored by TLC (SiO2, chloroform-methanol 10:1 mixture; stained with Dragendorff’s reagent). The reaction mixture was cooled in an ice bath, and excess of lithium aluminum hydride was quenched with water (5 mL). The ether layer was separated by decantation and residue was washed with ether (2 × 50 mL). The combined organic layers were dried with anhydrous Na2SO4, the solvent was evaporated in vacuum and the crude residue was stirred with chloroform (50 mL) and manganese dioxide (3.44 g, 40 mmol) at room temperature for 3 h. The reaction was monitored by TLC (SiO2, chloroform-methanol 10:1 mixture; stained with Dragendorff’s reagent). Manganese oxides were filtered off, precipitate was washed with chloroform (30 mL) and methanol (30 mL), the filtrate was evaporated in vacuum, and the residue was purified by column chromatography (SiO2, chloroform-methanol 15:1 mixture as an eluent, detected under UV lamp).
3-(5-tert-Butyl-2-ethyl-1-oxido-3,4-dihydro-2H-pyrrol-2-yl)-1-propanol (11a): 0.95 g, yield 42%. Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C13H25NO2)+: 227.1880; found:227.1882. IR (neat) νmax: 3346 (O–H), 1583 (C = N). 1H NMR (400 MHz; CDCl3, δ): 0.70 (t, Jt = 7.4 Hz, 3H), 1.17 (s, 9H, t-Bu), 1.22–1.34 (m, 1H), 1.34–1.51 (m, 3H), 1.73–1.89 (m, 4H), 2.45–2.51 (m, 2H), 3.46 (dt, Jt = 6.2 Hz, Jd = 1.8 Hz, 2H), 4.21 (br.s, 1H); 13C NMR (100 MHz; CDCl3, δ): 7.27, 23.63, 25.24, 26.05, 27.56, 30.98, 33.32, 33.53, 61.62, 80.12, 152.49.
3,3′-(5-tert-Butyl-1-oxido-3,4-dihydro-2H-pyrrole-2,2-diyl)di(1-propanol) (11b): 1.64 g, yield 64%. White crystalline solid, m.p. 136.7–137.2 °C (ethyl acetate). Elemental analysis: found: C, 65.33; H, 10.57; N, 5.44; calcd. for C14H27NO3: C, 65.53; H, 10.78; N, 5.51%; IR (KBr) νmax: 3344 (O–H), 1600 (C=N). 1H NMR (500 MHz; CDCl3, δ): 1.20 (s, 9H, t-Bu), 1.23–1.33 (m, 2H), 1.42–1.55 (m, 4H), 1.81–1.91 (m, 4H), 2.52–2.56 (m, 2H), 3.49 (t, Jt = 6.2 Hz, 4H), 4.38 (br.s, 2H); 13C NMR (125 MHz; CDCl3, δ): 24.19, 25.35, 26.02, 27.75, 33.50, 34.20, 61.60, 79.87, 154.38.
3.2.8. Reaction of Nitrones 11a and 11b with 2,2-Dimethoxypropane (General Method)
A mixture of 11a or 11b (10 mmol), 2,2-dimethoxypropane (48.9 mL, 400 mmol), PPTS (502 mg, 2 mmol), molecular sieves 4 Å (10 g) and anhydrous chloroform (50 mL) was stirred at room temperature for 48 h. The reaction was monitored by TLC (SiO2, chloroform-methanol 15:1 mixture; detected under UV lamp). After that, sieves were filtered off, and the filtrate was washed with aqueous sodium bicarbonate saturated solution (3 × 30 mL) and dried with anhydrous Na2SO4. The solvent was evaporated in vacuum, and the crude residue was purified by column chromatography (SiO2, chloroform-methanol 20:1 mixture as an eluent, detected under UV lamp) to give desired nitrone.
5-tert-Butyl-2-ethyl-2-[3-(1-methoxy-1-methylethoxy)propyl]-3,4-dihydro-2H-pyrrole 1-oxide (12a): 2.48 g, yield 83%. Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C17H33NO3)+:299.2455; found: 299.2465. IR (neat) νmax: 1578 (C = N). 1H NMR (500 MHz; CDCl3, δ): 0.75 (t, Jt = 7.4 Hz, 3H), 1.21 (s, 9H, t-Bu), 1.23 (s, 6H), 1.52 (dt, Jt = 13.2 Hz, Jd = 4.4 Hz, 1H), 1.77 (dt, Jt = 13.2 Hz, Jd = 4.4 Hz, 1H), 1.81–1.88 (m, 3H), 2.48–2.53 (m, 2H), 3.09 (s, 3H), 3.22–3.29 (m, 1H), 3.32–3.37 (m, 1H); 13C NMR (125 MHz; CDCl3, δ): 7.37, 23.69, 23.90, 24.20, 25.31, 27.48, 30.74, 33.30, 34.63, 48.21, 60.37, 79.89, 99.53, 150.58.
5-tert-Butyl-2,2-bis [3-(1-methoxy-1-methylethoxy)propyl]-3,4-dihydro-2H-pyrrole 1-oxide (12b): 3.2 g, yield 80%. Colorless oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C22H43NO5)+:401.3136; found: 401.3138.IR (neat) νmax: 1573 (C = N). 1H NMR (500 MHz; CDCl3, δ): 1.18 (s, 9H, t-Bu), 1.20 (s, 12H), 1.27–1.44 (m, 4H), 1.51 (dt, Jt = 12.8 Hz, Jd = 4.3 Hz, 2H), 1.75 (dt, Jt = 13.0 Hz, Jd = 4.5 Hz, 2H), 1.83–1.87 (m, 2H), 2.46–2.51 (m, 2H), 3.05 (s, 6H), 3.19–3.25 (m, 2H), 3.28–3.34 (m, 2H); 13C NMR (125 MHz; CDCl3, δ): 23.58, 24.13, 24.44, 25.21, 27.35, 33.21, 34.75, 48.13, 60.24, 79.29, 99.46, 150.31.
3.2.9. Reaction of Nitrones 12a and 12b with Ethyllithium (General Method)
A solution of ethyllithium in n-pentane (0.7 M, 70 mL) was slowly added dropwise to a solution of 12a or 12b (6 mmol) in dry benzene (10 mL) upon stirring under argon. The mixture was stirred for 20 h at room temperature, cooled in the ice bath, and quenched with water (20 mL). The organic layer was separated and water layer was extracted with diethyl ether (2 × 30 mL). Combined extracts were dried with Na2SO4 and the solvent was evaporated in vacuum. The crude residue was dissolved in methanol (70 mL) and basified with aqueous solution of sodium hydroxide (1 M, 20 mL). Methylene blue (6 mg, 0.02 mmol) was added to the mixture, and the air was bubbled until the solution had turned dark blue. The methanol was distilled off in vacuum, and the residue was extracted with diethyl ether (3 × 30 mL). Combined extract was dried with Na2SO4 and the solvent was evaporated in vacuum. The residue was dissolved in the 30 mL of mixture methanol:water (1:1), and PPTS (500 mg) was added. The mixture was stirred for 1 h at room temperature, methanol was distilled off in vacuum and residue was extracted with diethyl ether (3 × 30 mL). Combined extract was dried with Na2SO4 and the solvent was evaporated in vacuum. The residue was purified by column chromatography (SiO2, chloroform-methanol 30:1 or hexane-ethyl acetate1:1 mixture as an eluent, detected under UV lamp) to give desired nitroxide.
2-tert-Butyl-2,5-diethyl-5-(3-hydroxypropyl)pyrrolidine-1-oxyl (14a):yield 1.04 g (68%), yellow oil. HRMS (EI/DFS) m/z [M]+ calcd. for (C15H30NO2)+: 256.2271; found: 256.2273. IR (neat) νmax: 3425 (O–H). 1H NMR (400 MHz; CD3OD/CDCl3, Zn/CF3COOH system δ): 0.74 (q, Jq = 7.3 Hz, 3H), 0.85 (t, Jt = 7.5 Hz, 3H, signals of the first isomer), 0.87 (s, 9H, t-Bu), 0.88 (t, Jt = 7.5 Hz, 3H, signals of the second isomer), 1.37–1.99 (m, 12H), 3.24–3.37 (m, 1H), 3.44–3.55 (m, 1H).
2-tert-Butyl-2-ethyl-5,5-bis(3-hydroxypropyl)pyrrolidine-1-oxyl (14b):yield 0.91 g (53%), yellow crystalline solid,. m.p. 93.0–95.2 °C (hexane-ethyl acetate 1:1). Found: C, 67.44; H, 11.19; N, 5.06; calcd. for C16H32NO3: C, 67.09; H, 11.26; N, 4.89%; IR (KBr) νmax: 3332 (O–H). 1H NMR (400 MHz; CD3OD/CDCl3, Zn/CF3COOH system δ): 0.77 (t, Jt = 7.5 Hz, 3H), 0.78 (s, 9H, t-Bu), 1.20–1.37 (m, 4H), 1.40–1.62 (m, 4H), 1.62–1.74 (m, 4H), 1.74–1.93 (m, 2H), 3.22–3.42 (m, 4H).


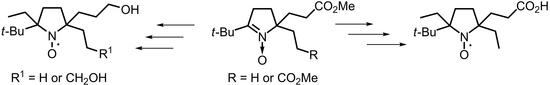
 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}