
Greener Synthesis of Antiproliferative Furoxans via Multicomponent Reactions

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
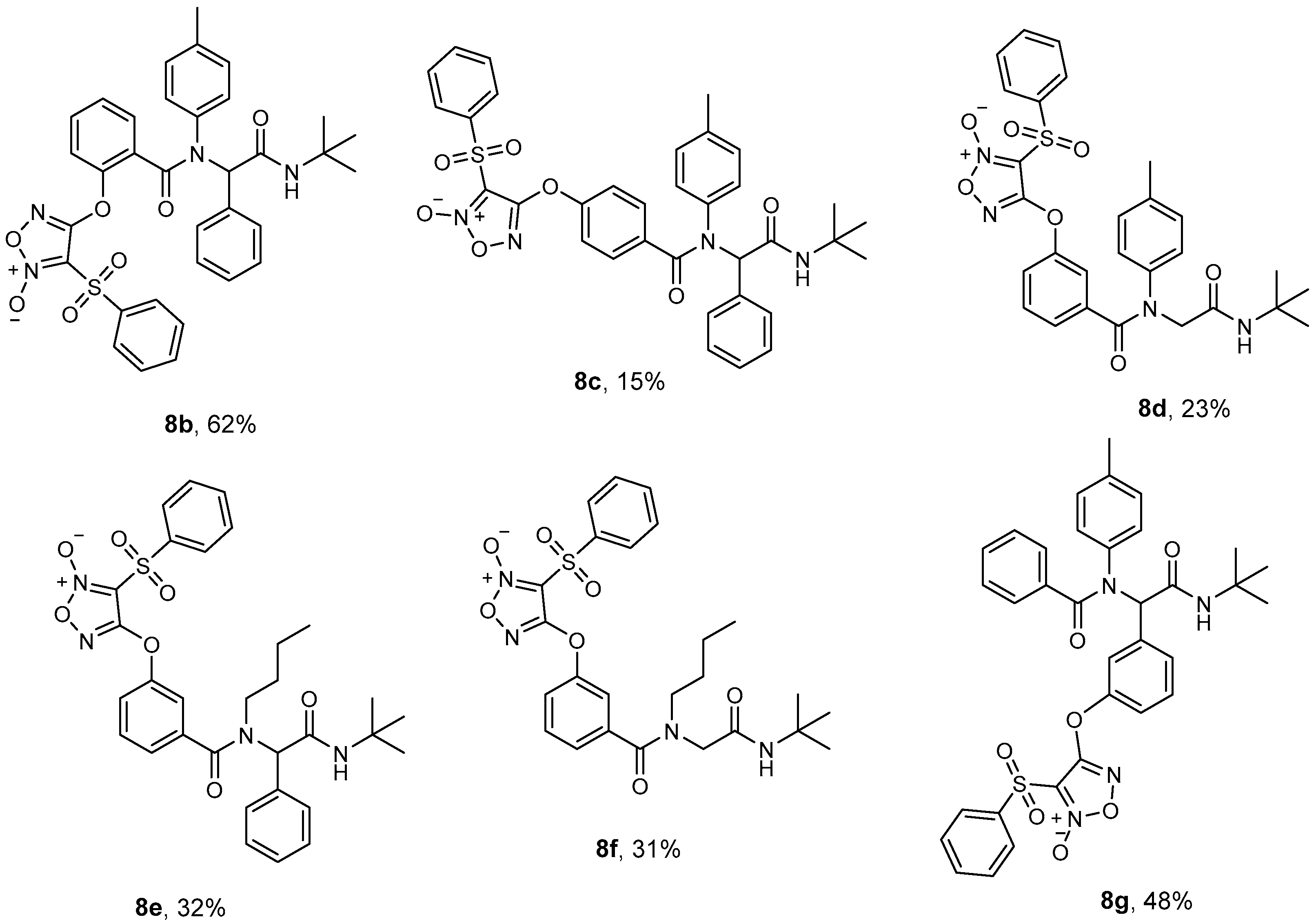
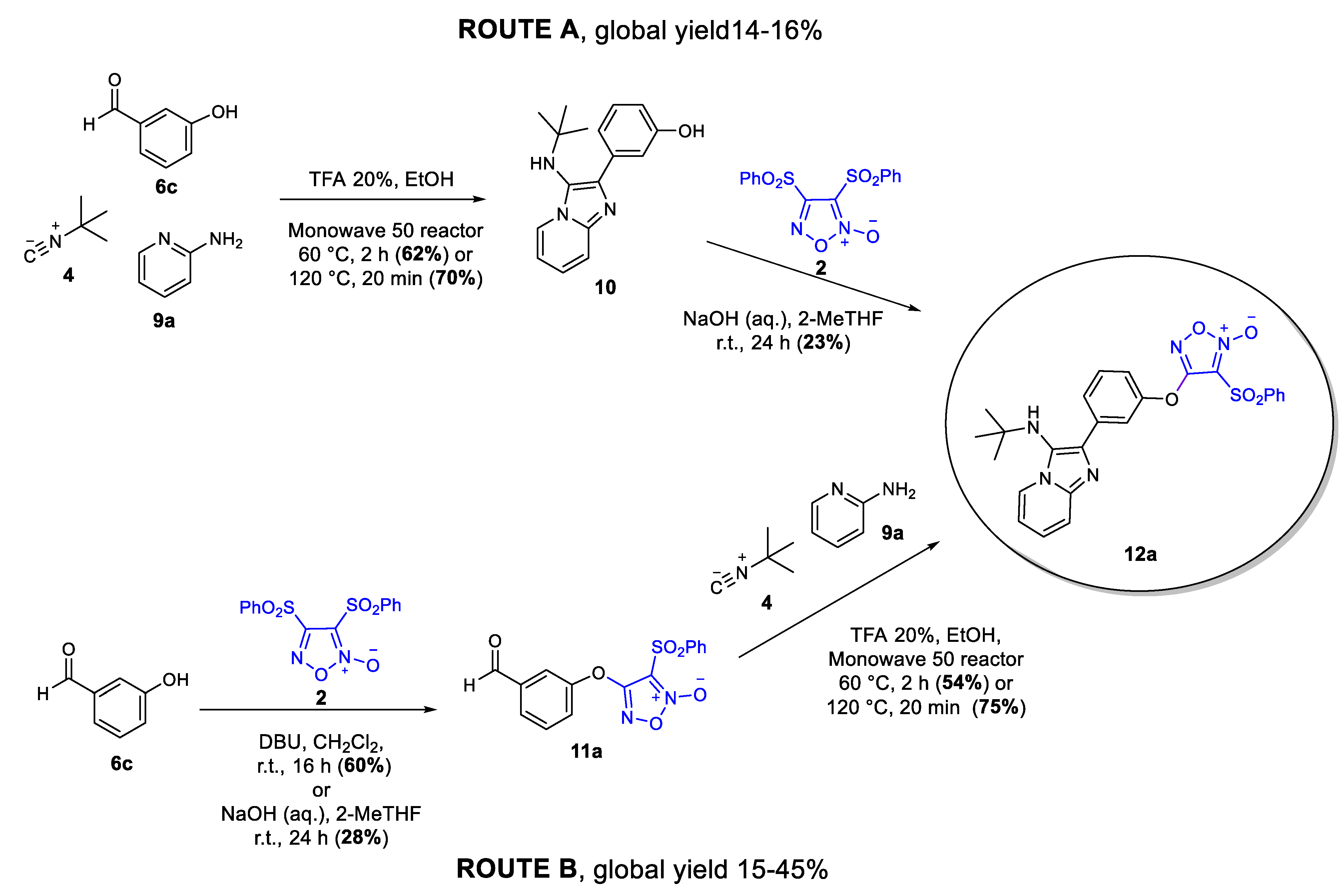
2.1.1. Ugi Reaction
2.1.2. GBB Reaction
2.2. Biology
2.2.1. Antiproliferative Activity
2.2.2. Selectivity Index
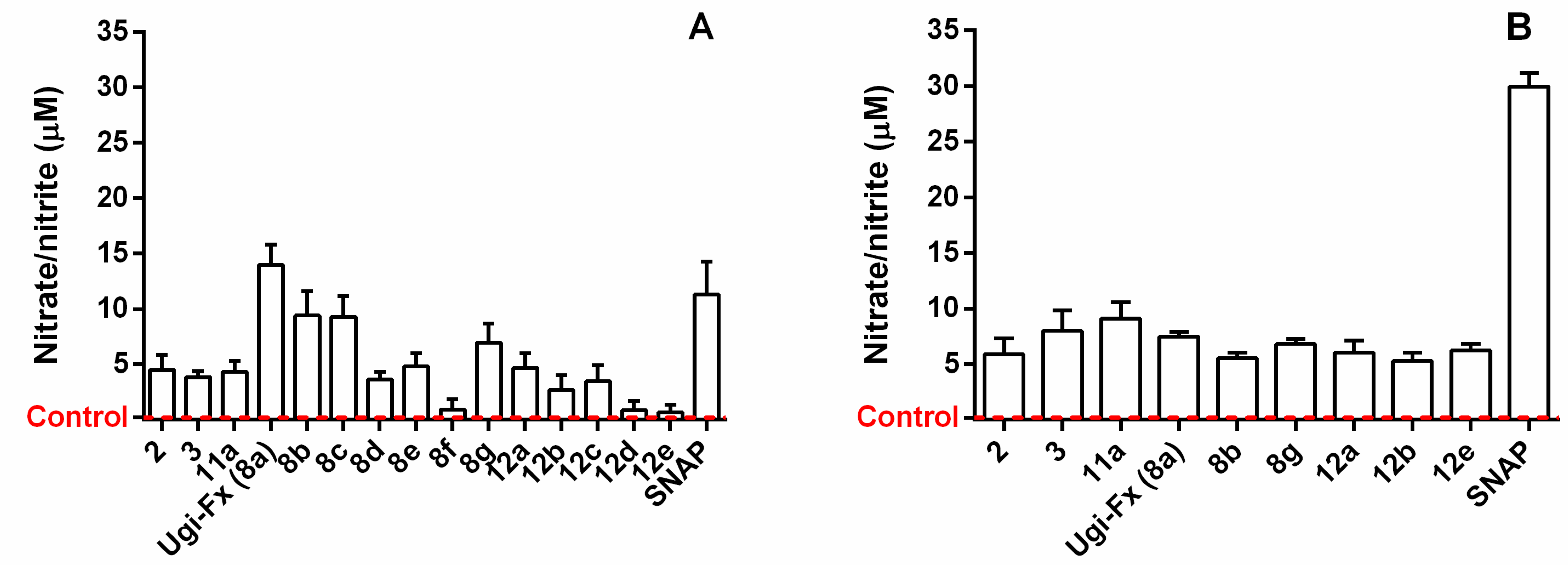
2.2.3. Assessment of ·NO Release
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental Information
3.1.2. Experimental Procedures and Characterization Data for the Compounds
3.2. Biology
3.2.1. Materials
3.2.2. Cell Culture
3.2.3. Antiproliferative Activity
3.2.4. Nitric-Oxide-Releasing Activity
3.2.5. Antiproliferative Activity with Hemoglobin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization (WHO). Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 30 December 2021).
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmström, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Zhao, T.; Kang, D.; Zhang, J.; Song, Y.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414. [Google Scholar] [CrossRef]
- Bryan, M.C.; Dillon, B.; Hamann, L.G.; Hughes, G.J.; Kopach, M.E.; Peterson, E.A.; Pourashraf, M.; Raheem, I.; Richardson, P.; Richter, D.; et al. Sustainable Practices in Medicinal Chemistry: Current State and Future Directions. J. Med. Chem. 2013, 56, 6007–6021. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Chandgude, A.L.; Dömling, A. Multicomponent Reactions, Union of MCRs and Beyond. Chem. Rec. 2015, 15, 981–996. [Google Scholar] [CrossRef]
- Ruijter, E.; Orru, R.; Synthetic and BioOrganic Chemistry Group. Multicomponent reactions in drug discovery and medicinal chemistry. Drug Discov. Today Technol. 2018, 29, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Ingold, M.; Dapueto, R.; Victoria, S.; Galliusi, G.; Batthyàny, C.; Bollati-Fogolín, M.; Tejedor, D.; García-Tellado, F.; Padrón, J.M.; Porcal, W.; et al. A green multicomponent synthesis of tocopherol analogues with antiproliferative activities. Eur. J. Med. Chem. 2018, 143, 1888–1902. [Google Scholar] [CrossRef] [Green Version]
- Ingold, M.; Colella, L.; Hernández, P.; Batthyány, C.; Tejedor, D.; Puerta, A.; García-Tellado, F.; Padrón, J.M.; Porcal, W.; López, G.V. A Focused Library of NO-Donor Compounds with Potent Antiproliferative Activity Based on Green Multicomponent Reactions. ChemMedChem 2019, 14, 1669–1683. [Google Scholar] [CrossRef] [Green Version]
- Ugi, I.; Meyr, R.; Fetzer, U.; Steinbrückner, C. Versuche mit Isonitrilen. Angew. Chem. 1959, 71, 386–388. [Google Scholar]
- Groebke, K.; Weber, L.; Mehlin, F. Synthesis of imidazo[1,2-a] annulated pyridines, pyrazines and pyrimidines by a novel three-component condensation. Synlett 1998, 1998, 661–663. [Google Scholar] [CrossRef]
- Blackburn, C.; Guan, B.; Fleming, P.; Shiosaki, K.; Tsai, S. Parallel synthesis of 3-aminoimidazo[1,2-a]pyridines and pyrazines by a new three-component condensation. Tetrahedron Lett. 1998, 39, 3635–3638. [Google Scholar] [CrossRef]
- Bienayme, H.; Bouzid, K. A New Heterocyclic Multicomponent Reaction For the Combinatorial Synthesis of Fused 3-Aminoimidazoles. Angew. Chem. Int. Ed. 1998, 37, 2234–2237. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Dömling, A. Modern multicomponent reactions for better drug syntheses. Org. Chem. Front. 2014, 1, 834–837. [Google Scholar] [CrossRef]
- Boltjes, A.; Dömling, A. The Groebke-Blackburn-Bienaymé Reaction. Eur. J. Org. Chem. 2019, 2019, 7007–7049. [Google Scholar] [CrossRef]
- Pirrung, M.C.; Sarma, K. Das Aqueous medium effects on multi-component reactions. Tetrahedron 2005, 61, 11456–11472. [Google Scholar] [CrossRef]
- Vidyacharan, S.; Shinde, A.H.; Satpathi, B.; Sharada, D.S. A facile protocol for the synthesis of 3-aminoimidazo-fused heterocycles via the Groebke-Blackburn-Bienayme reaction under catalyst-free and solvent-free conditions. Green Chem. 2014, 16, 1168–1175. [Google Scholar] [CrossRef]
- Dolzhenko, A.V. Microwave-Assisted Multicomponent Reactions; Elsevier: Amsterdam, The Netherlands, 2021; ISBN 9780128198483. [Google Scholar]
- Jovanović, M.; Zhukovsky, D.; Podolski-Renić, A.; Domračeva, I.; Žalubovskis, R.; Senćanski, M.; Glišić, S.; Sharoyko, V.; Tennikova, T.; Dar’in, D.; et al. Novel electrophilic amides amenable by the Ugi reaction perturb thioredoxin system via thioredoxin reductase 1 (TrxR1) inhibition: Identification of DVD-445 as a new lead compound for anticancer therapy. Eur. J. Med. Chem. 2019, 181, 111580. [Google Scholar] [CrossRef]
- Fouad, M.A.; Hamida Abdel-Hamida, H.; Salah Ayoup, M.S. Two decades of recent advances of Ugi reactions: Synthetic and pharmaceutical applications. RSC Adv. 2020, 10, 42644–42681. [Google Scholar] [CrossRef]
- Serafim, R.A.M.; Pernichelle, F.G.; Ferreira, E.I. The latest advances in the discovery of nitric oxide hybrid drug compounds. Expert Opin. Drug Discov. 2017, 12, 941–953. [Google Scholar] [CrossRef]
- Glynn, S.A. Emerging novel mechanisms of action for nitric oxide in cancer progression. Curr. Opin. Physiol. 2019, 9, 18–25. [Google Scholar] [CrossRef]
- Seabra, A.B.; Durán, N. Nitric oxide donors for prostate and bladder cancers: Current state and challenges. Eur. J. Pharmacol. 2018, 826, 158–168. [Google Scholar] [CrossRef] [PubMed]
- Fershtat, L.L.; Makhova, N.N. Molecular Hybridization Tools in the Development of Furoxan-Based NO-Donor Prodrugs. ChemMedChem 2017, 12, 622–638. [Google Scholar] [CrossRef] [PubMed]
- Ramazani, A.; Karimi, M.; Hosseinzadeh, Z.; Rezayati, S.; Hanifehpour, Y.; Joo, S.W. Syntheses and Antitumor Properties of Furoxan Derivatives. Curr. Org. Chem. 2021, 25, 757–778. [Google Scholar] [CrossRef]
- Huang, Z.; Fu, J.; Zhang, Y. Nitric Oxide Donor-Based Cancer Therapy: Advances and Prospects. J. Med. Chem. 2017, 60, 7617–7635. [Google Scholar] [CrossRef]
- Vannini, F.; MacKessack-Leitch, A.C.; Eschbach, E.K.; Chattopadhyay, M.; Kodela, R.; Kashfi, K. Synthesis and anti-cancer potential of the positional isomers of NOSH-aspirin (NBS-1120) a dual nitric oxide and hydrogen sulfide releasing hybrid. Bioorg. Med. Chem. Lett. 2015, 25, 4677–4682. [Google Scholar] [CrossRef] [Green Version]
- Kappe, C.O.; Account, P. My Twenty Years in Microwave Chemistry: From Kitchen Ovens to Microwaves that aren’t Microwaves. Chem. Rec. 2019, 19, 15–39. [Google Scholar] [CrossRef]
- Pace, V.; Hoyos, P.; Castoldi, L.; Domínguez De María, P.; Alcántara, A.R. 2-Methyltetrahydrofuran (2-MeTHF): A Biomass-Derived Solvent with Broad Application in Organic Chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef]
- Přibylka, A.; Krchňák, V.; Schütznerová, E. Environmentally friendly SPPS I. Application of NaOH in 2-MeTHF/methanol for Fmoc removal. Green Chem. 2019, 21, 775–779. [Google Scholar] [CrossRef]
- dos Santos Fernandes, G.F.; de Souza, P.C.; Marino, L.B.; Chegaev, K.; Guglielmo, S.; Lazzarato, L.; Fruttero, R.; Chung, M.C.; Pavan, F.R.; dos Santos, J.L. Synthesis and biological activity of furoxan derivatives against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2016, 123, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Vichai, V.; Kirtikara, K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006, 1, 1112–1116. [Google Scholar] [CrossRef]
- Pérez, F.; Varela, M.; Canclini, L.; Acosta, S.; Martínez-López, W.; López, G.V.; Hernández, P. Furoxans and tocopherol analogs-furoxan hybrids as anticancer agents. Anticancer Drugs 2019, 30, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Burov, O.N.; Kletskii, M.E.; Fedik, N.S.; Lisovin, A.V.; Kurbatov, S.V. Mechanism of Thiol-Induced Nitrogen(II) Oxide Donation by Furoxans: A Quantum-Chemical Study. Chem. Heterocycl. Compd. 2016, 51, 951–960. [Google Scholar] [CrossRef]
- Sun, J.; Zhang, X.; Broderick, M.; Fein, H. Measurement of Nitric Oxide Production in Biological Systems by Using Griess Reaction Assay. Sensors 2003, 3, 276–284. [Google Scholar] [CrossRef] [Green Version]
- Yu, N.; Li, N.; Wang, K.; Deng, Q.; Lei, Z.; Sun, J.; Chen, L. Design, synthesis and biological activity evaluation of novel scopoletin-NO donor derivatives against MCF-7 human breast cancer in vitro and in vivo. Eur. J. Med. Chem. 2021, 224, 113701. [Google Scholar] [CrossRef]
- Moharram, S.; Zhou, A.; Wiebe, L.I.; Knaus, E.E. Design and Synthesis of 3′- and 5′-O-(3-Benzenesulfonylfuroxan-4-yl)-2′-deoxyuridines: Biological Evaluation as Hybrid Nitric Oxide Donor−Nucleoside Anticancer Agents. J. Med. Chem. 2004, 47, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Boiani, M.; Cerecetto, H.; González, M. Cytotoxicity of furoxans: Quantitative structure-activity relationships study. Il Farmaco 2004, 59, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Cerecetto, H.; Porcal, W. Pharmacological properties of furoxans and benzofuroxans: Recent developments. Mini-Rev. Med. Chem. 2005, 5, 57–71. [Google Scholar] [CrossRef]
- Sodano, F.; Gazzano, E.; Rolando, B.; Marini, E.; Lazzarato, L.; Fruttero, R.; Riganti, C.; Gasco, A. Tuning NO release of organelle-targeted furoxan derivatives and their cytotoxicity against lung cancer cells. Bioorg. Chem. 2021, 111, 104911. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| A549 (Lung) | HBL-100 (Breast) | HeLa (Cervix) | SW1573 (Lung) | T-47D (Breast) | WiDr (Colon) | |
|---|---|---|---|---|---|---|
| GI50 (µM) | 2.3 (±0.5) | 0.23 (±0.02) | 1.8 (±0.3) | 0.021 (±0.013) | 1.9 (±0.3) | 3.0 (±0.6) |
| SI | 4.3 | 43 | 5.5 | 471.4 | 5.2 | 3.3 |
| Compound | Cell Line | Compound | Cell Line | ||
|---|---|---|---|---|---|
| T24 | LNCaP | T24 | LNCaP | ||
| Furoxan reference compounds | |||||
| 3 | 8.60 (±1.68) | 32.46 (±9.96) | 11a | 7.08 (±1.70) | 24.52 (±5.20) |
| Ugi products | GBB products | ||||
| 7a | >100 | >100 | 10 | >100 | >100 |
| Ugi-Fx (8a) | 0.73 (±0.12) | 20.2 (±4.66) | 12a | 1.76 (±0.34) | 24.7 (±1.83) |
| 8b | 1.83 (±0.59) | 13.3 (±5.60) | 12b | 0.85 (±0.25) | 7.70 (±2.13) |
| 8c | 2.18 (±0.63) | 18.8 (±1.62) | 12c | 2.99 (±0.98) | 24.2 (±3.40) |
| 8d | 2.10 (±0.45) | 21.79 (±8.39) | 12d | 2.72 (±0.41) | 8.13 (±1.55) |
| 8e | 1.53 (±0.29) | 21.82 (±6.09) | 12e | 0.83 (±0.14) | 16.53 (±4.99) |
| 8f | 2.86 (±0.46) | 29.87 (±6.96) | Positive control | ||
| 8g | 1.70 (±0.46) | 36.7 (±11.8) | Cisplatin | 3.28 (±1.20) | 20.3 (±4.20) |
| Compound | HaCaT GI50 (µM) a | SIb | |
|---|---|---|---|
| T24 | LNCaP | ||
| 2 | 1.47 (±1.11) c | 1.1c | ND |
| 3 | 14.40 (±1.92) | 1.7 | 0.4 |
| 11a | 7.83 (±1.43) | 1.1 | 0.3 |
| Ugi-Fx (8a) | 10.58 (±3.08) | 14.5 | 0.5 |
| 8b | 3.78 (±1.15) | 2.1 | 0.3 |
| 8c | 4.62 (±0.99) | 2.1 | 0.2 |
| 8d | 9.99 (±2.70) | 4.8 | 0.5 |
| 8e | 11.78 (±2.57) | 4.5 | 0.5 |
| 8f | 11.85 (±2.73) | 7.7 | 0.4 |
| 8g | 4.99 (±1.19) | 2.9 | 0.1 |
| 12a | 7.44 (±2.13) | 4.2 | 0.3 |
| 12b | 3.11 (±0.94) | 3.7 | 0.4 |
| 12c | 5.22 (±1.20) | 1.7 | 0.2 |
| 12d | 9.84 (±2.06) | 3.6 | 1.2 |
| 12e | 9.01 (±2.06) | 10.8 | 0.2 |
| Cisplatin | 2.51 (±1.19) | 0.8 | 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ingold, M.; de la Sovera, V.; Dapueto, R.; Hernández, P.; Porcal, W.; López, G.V. Greener Synthesis of Antiproliferative Furoxans via Multicomponent Reactions. Molecules 2022, 27, 1756. https://doi.org/10.3390/molecules27061756
Ingold M, de la Sovera V, Dapueto R, Hernández P, Porcal W, López GV. Greener Synthesis of Antiproliferative Furoxans via Multicomponent Reactions. Molecules. 2022; 27(6):1756. https://doi.org/10.3390/molecules27061756
Chicago/Turabian StyleIngold, Mariana, Victoria de la Sovera, Rosina Dapueto, Paola Hernández, Williams Porcal, and Gloria V. López. 2022. "Greener Synthesis of Antiproliferative Furoxans via Multicomponent Reactions" Molecules 27, no. 6: 1756. https://doi.org/10.3390/molecules27061756