
Catalyst-Free Site Selective Hydroxyalkylation of 5-Phenylthiophen-2-amine with α-Trifluoromethyl Ketones through Electrophilic Aromatic Substitution





Abstract
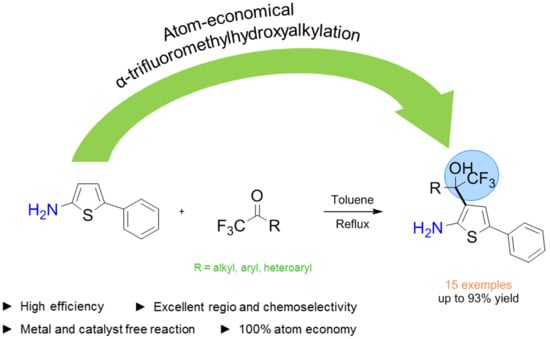
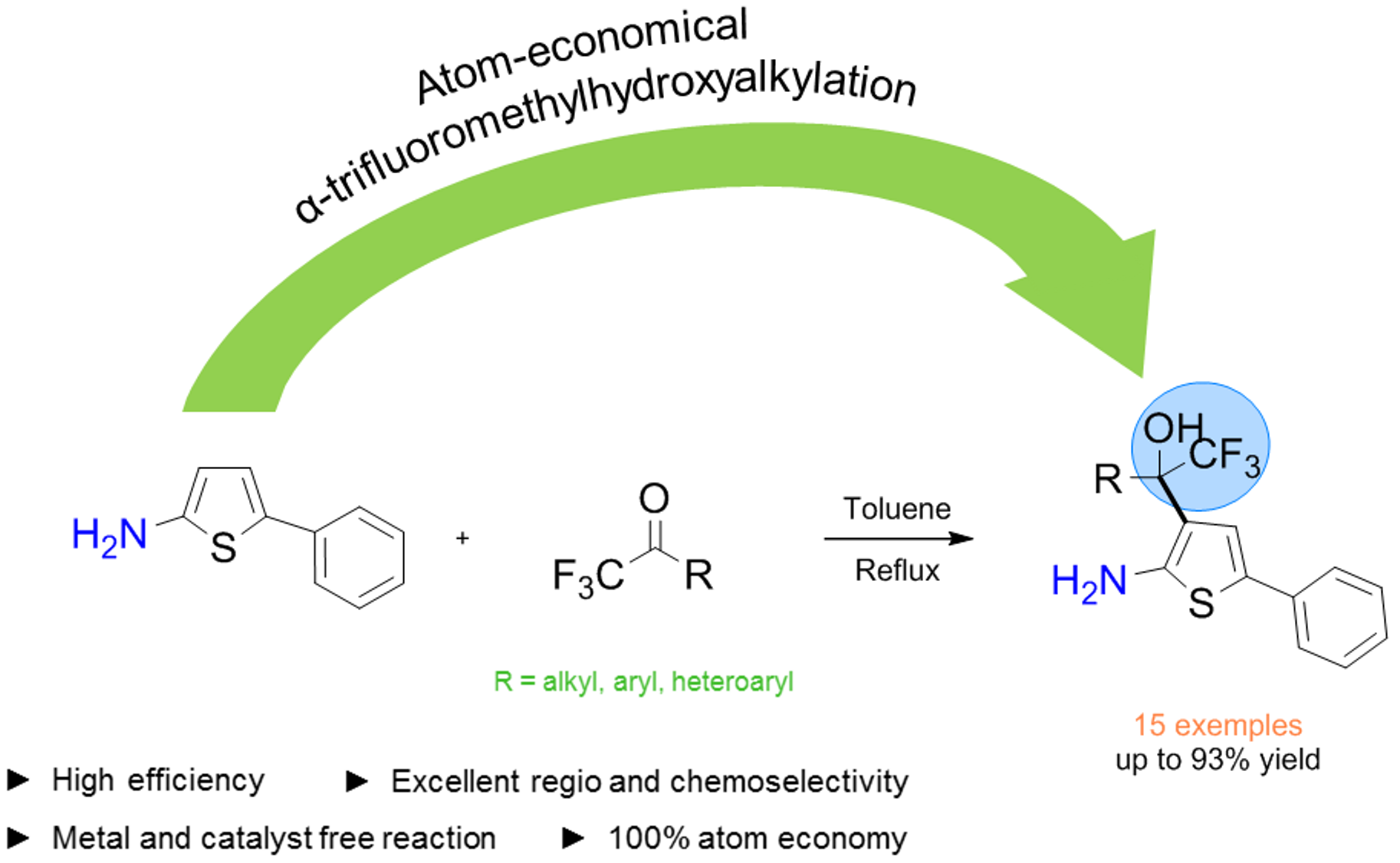
:
1. Introduction
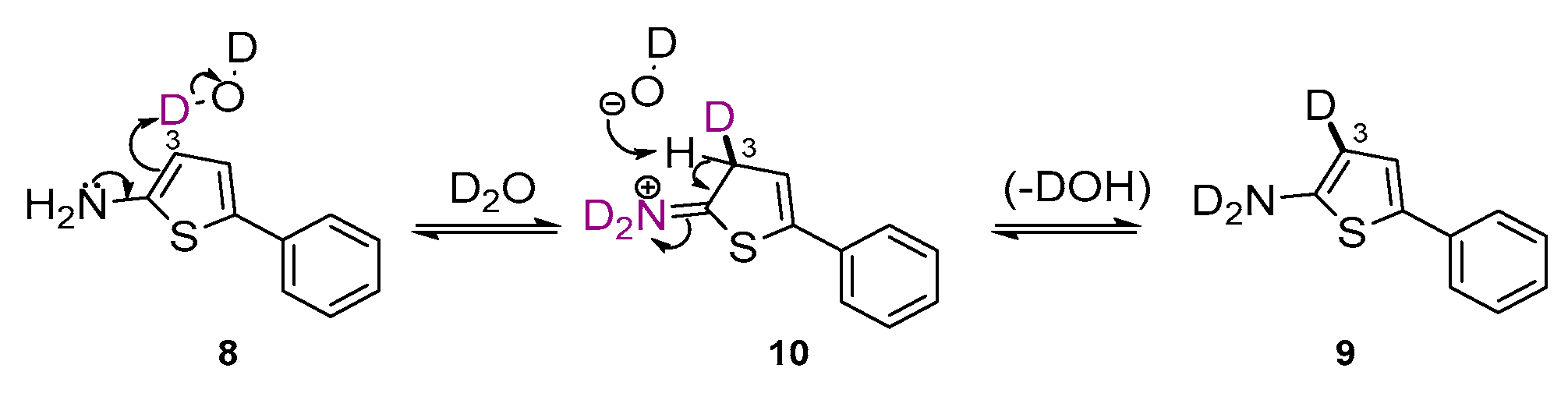
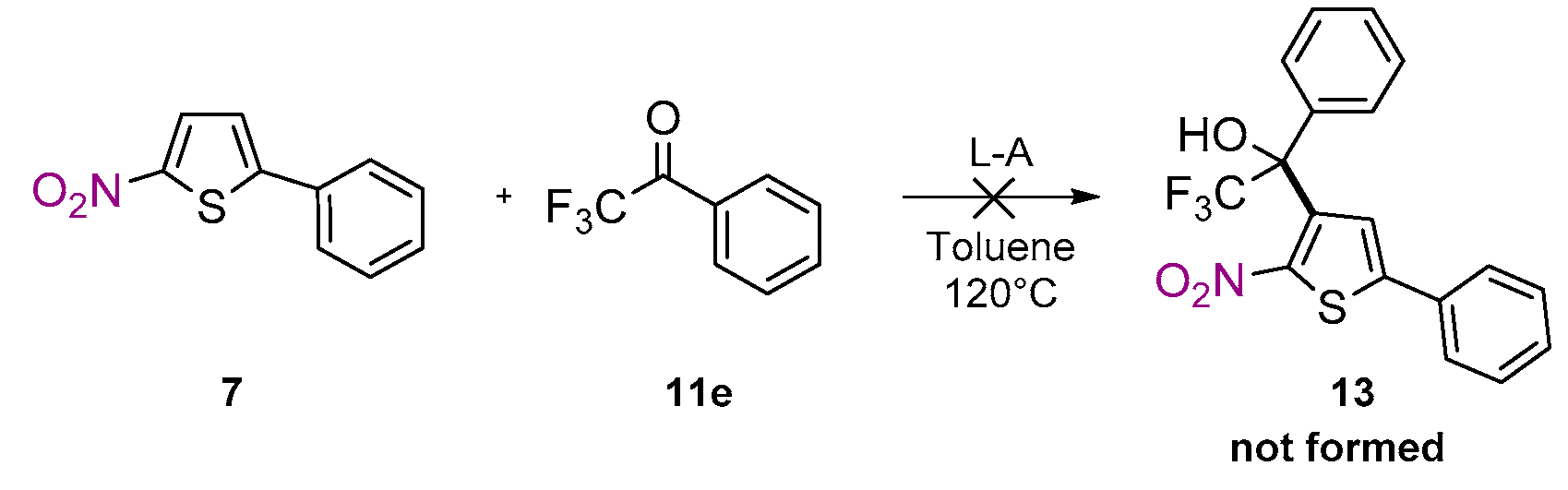
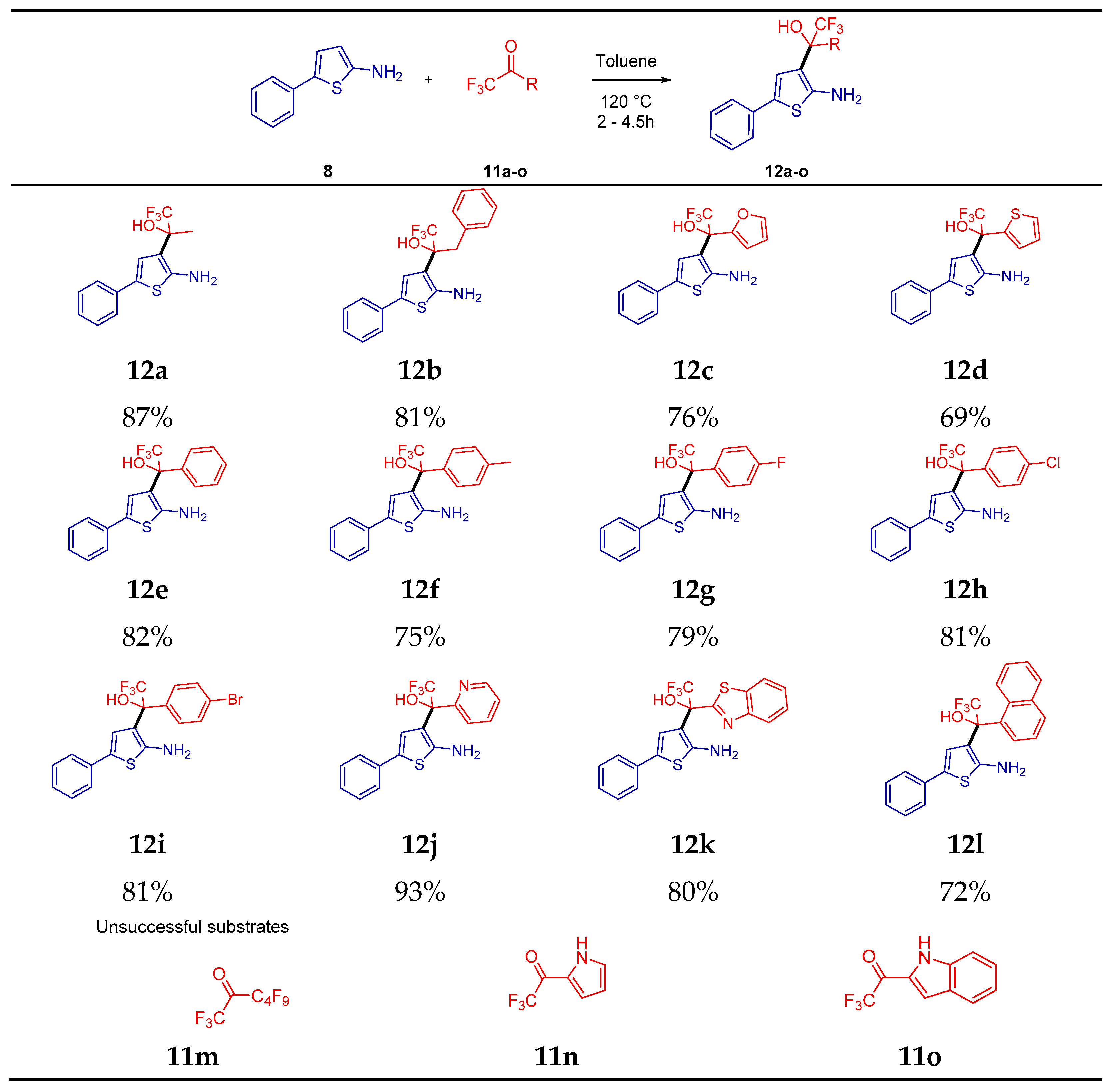
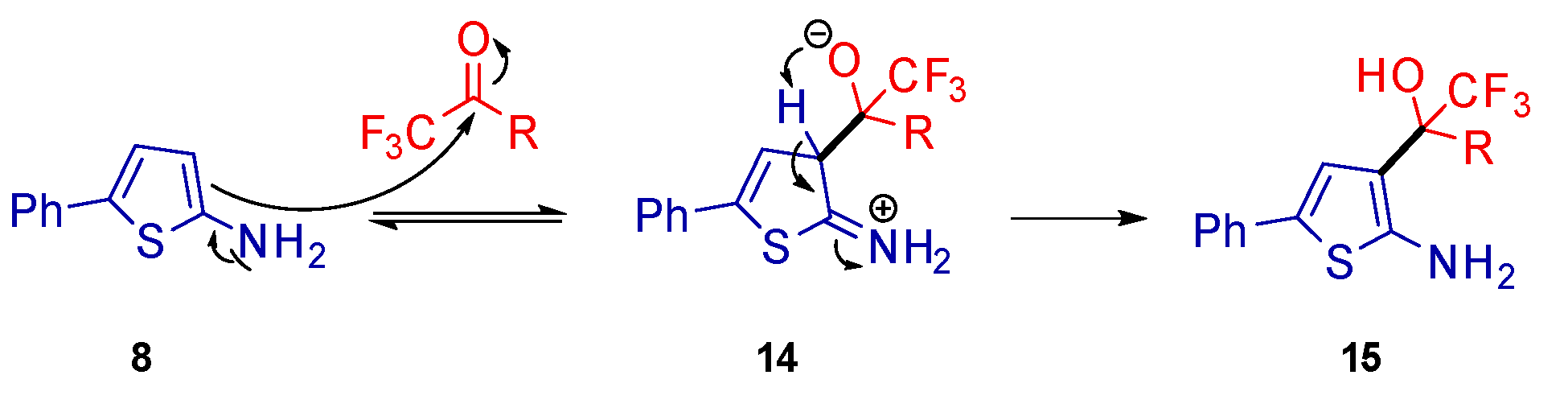
2. Results
3. Conclusions
4. Materials and Methods
4.1. General Experimental Methods
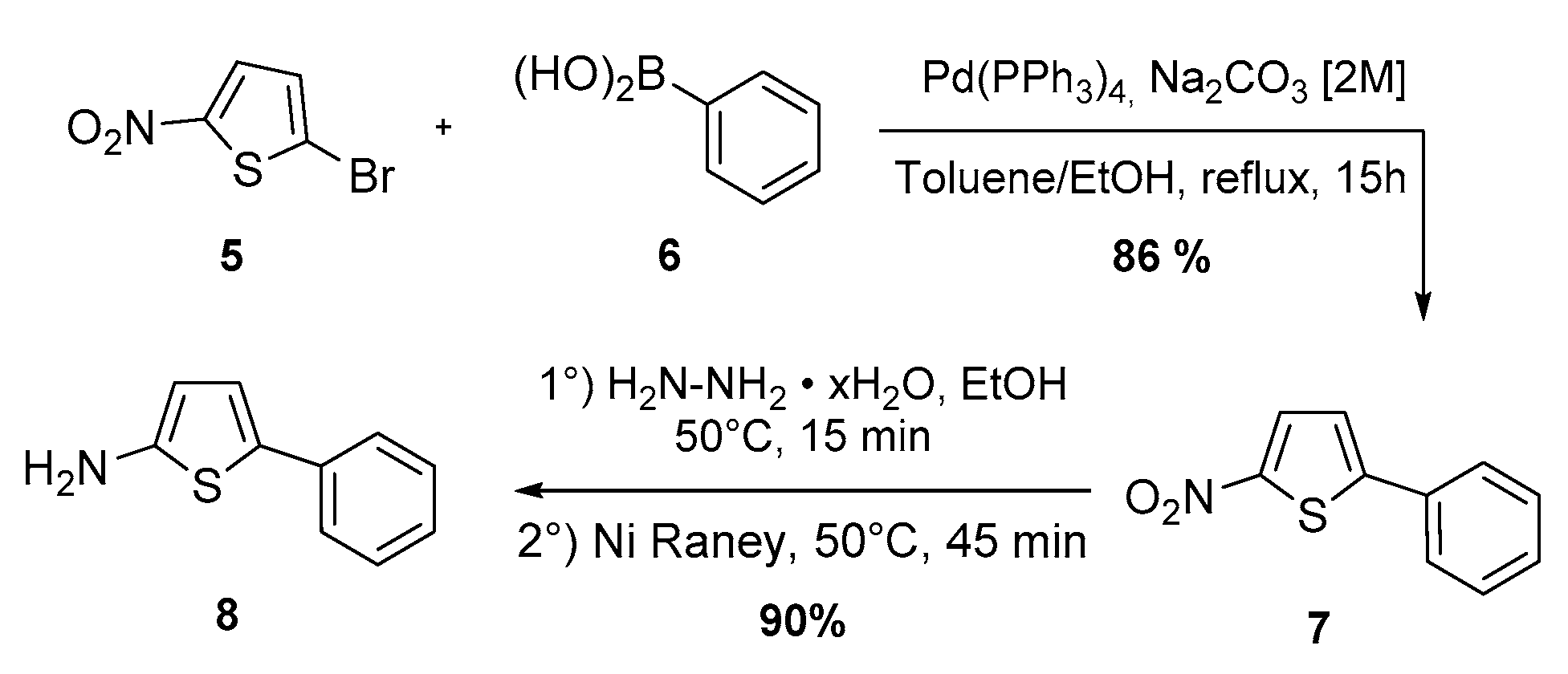
4.2. Preparation of 5-Phenylthiophen-2-amine (8)
4.3. Procedure for the Preparation of Trifluorohydroxyalkyl-5-Phenylthiophen-2-amine (12a–12l)
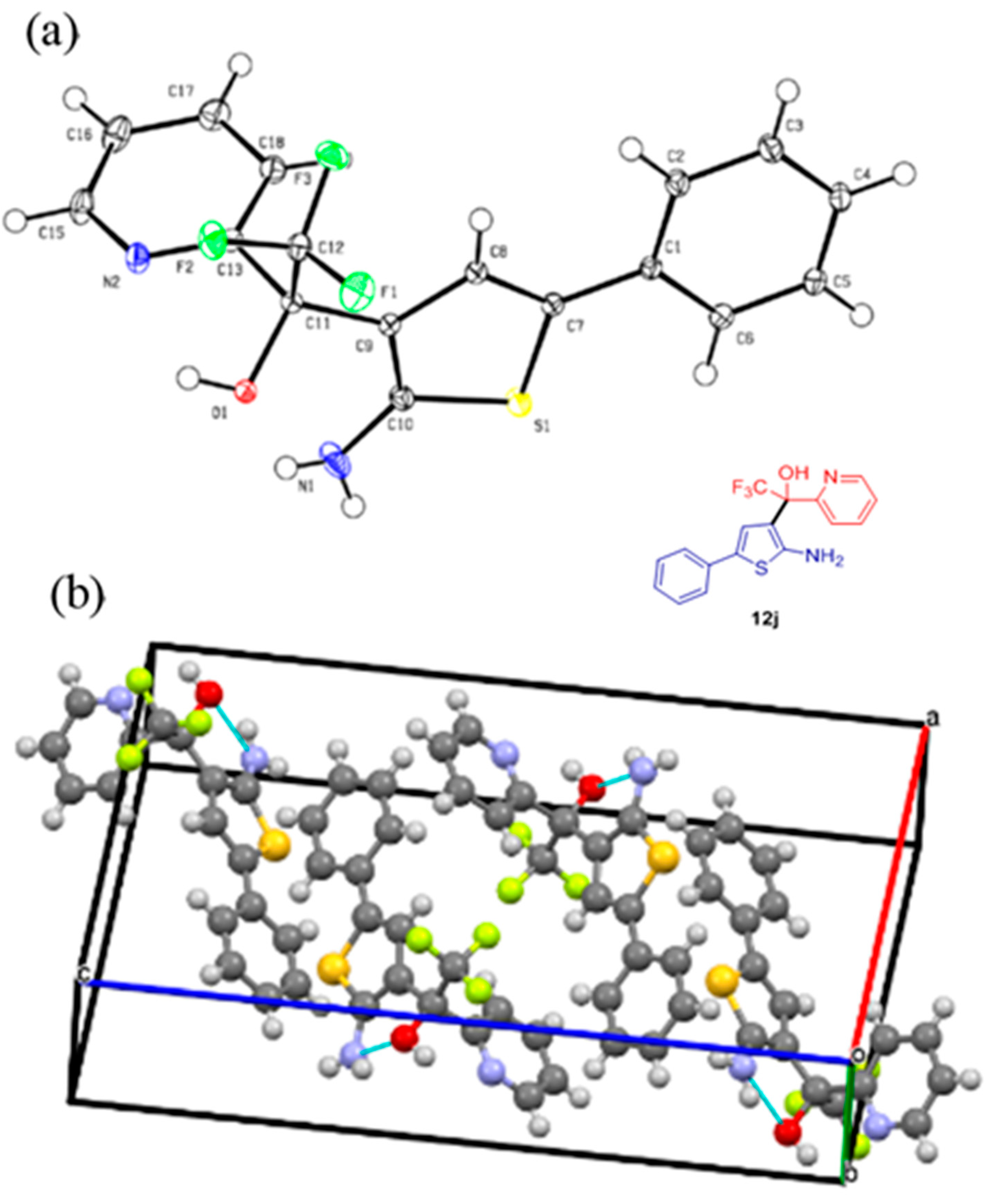
4.4. Crystallographic Data
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bozorov, K.; Nie, L.F.; Zhao, J.; Aisa, H.A. 2-Aminothiophene scaffolds: Diverse biological and pharmacological attributes in medicinal chemistry. Eur. J. Med. Chem. 2017, 140, 465–493. [Google Scholar] [CrossRef]
- Thanna, S.; Knudson, S.E.; Grzegorzewicz, A.; Kapil, S.; Goins, C.M.; Ronning, D.R.; Jackson, M.; Slayden, R.A.; Sucheck, S.J. Synthesis and evaluation of new 2-aminothiophenes against: Mycobacterium tuberculosis. Org. Biomol. Chem. 2016, 14, 6119–6133. [Google Scholar] [CrossRef]
- Scheich, C.; Puetter, V.; Schade, M. Novel small molecule inhibitors of MDR mycobacterium tuberculosis by NMR fragment screening of antigen 85C. J. Med. Chem. 2010, 53, 8362–8367. [Google Scholar] [CrossRef]
- Oza, V.; Ashwell, S.; Almeida, L.; Brassil, P.; Breed, J.; Deng, C.; Gero, T.; Grondine, M.; Horn, C.; Ioannidis, S.; et al. Discovery of checkpoint kinase inhibitor (S)-5-(3-fluorophenyl)-N-(piperidin-3-yl)-3-ureidothiophene-2-carboxamide (AZD7762) by structure-based design and optimization of thiophenecarboxamide ureas. J. Med. Chem. 2012, 55, 5130–5142. [Google Scholar] [CrossRef]
- Desantis, J.; Nannetti, G.; Massari, S.; Barreca, M.L.; Manfroni, G.; Cecchetti, V.; Palù, G.; Goracci, L.; Loregian, A.; Tabarrini, O. Exploring the cycloheptathiophene-3-carboxamide scaffold to disrupt the interactions of the influenza polymerase subunits and obtain potent anti-influenza activity. Eur. J. Med. Chem. 2017, 138, 128–139. [Google Scholar] [CrossRef]
- Narlawar, R.; Lane, J.R.; Doddareddy, M.; Lin, J.; Brussee, J.; Ijzerman, A.P. Hybrid ortho/allosteric ligands for the adenosine A1 receptor. J. Med. Chem. 2010, 53, 3028–3037. [Google Scholar] [CrossRef]
- Tang, J.; Huber, A.D.; Pineda, D.L.; Boschert, K.N.; Wolf, J.J.; Kankanala, J.; Xie, J.; Sarafianos, S.G.; Wang, Z. 5-Aminothiophene-2,4-dicarboxamide analogues as hepatitis B virus capsid assembly effectors. Eur. J. Med. Chem. 2019, 164, 179–192. [Google Scholar] [CrossRef]
- Aly, H.M.; Saleh, N.M.; Elhady, H.A. Design and synthesis of some new thiophene, thienopyrimidine and thienothiadiazine derivatives of antipyrine as potential antimicrobial agents. Eur. J. Med. Chem. 2011, 46, 4566–4572. [Google Scholar] [CrossRef]
- Campaigne, E.; Foye, W.O. The synthesis of 2,5-diarylthiophenes. J. Org. Chem. 1952, 17, 1405–1412. [Google Scholar] [CrossRef]
- Gewald, K. Heterocyclen aus CH-aciden Nitrilen, VIII. 2-Amino-thiophene aus methylenaktiven Nitrilen; Carbonylverbindungen und Schwefel. Chem. Ber. 1965, 98, 3571–3577. [Google Scholar] [CrossRef]
- Minetto, G.; Raveglia, L.F.; Sega, A.; Taddei, M. Microwave-assisted Paal-Knorr reaction—Three-step regiocontrolled synthesis of polysubstituted furans, pyrroles and thiophenes. Eur. J. Org. Chem. 2005, 2005, 5277–5288. [Google Scholar] [CrossRef]
- Revelant, G.; Dunand, S.; Hesse, S.; Kirsch, G. Microwave-assisted synthesis of 5-substituted 2-aminothiophenes starting from arylacetaldehydes. Synthesis 2011, 2011, 2935–2940. [Google Scholar] [CrossRef]
- Gouda, M.A.; Al-Ghorbani, M.; Al-Zaqri, N. Synthesis and cytotoxic activity of some new heterocycles incorporating cyclohepta[b]thiophene-3-carboxamide derivatives. J. Heterocycl. Chem. 2020, 57, 3664–3672. [Google Scholar] [CrossRef]
- Ibrahim, B.A.; Mohareb, R.M. Uses of ethyl benzoyl acetate for the synthesis of thiophene, pyran, and pyridine derivatives with antitumor activities. J. Heterocycl. Chem. 2020, 57, 4023–4035. [Google Scholar] [CrossRef]
- Hwang, J.; Borgelt, L.; Wu, P. Multicomponent petasis reaction for the synthesis of functionalized 2-aminothiophenes and thienodiazepines. ACS Comb. Sci. 2020, 22, 495–499. [Google Scholar] [CrossRef]
- Touré, B.B.; Hall, D.G. Natural product synthesis using multicomponent reaction strategies. Chem. Rev. 2009, 109, 4439–4486. [Google Scholar] [CrossRef]
- Bauer, I.; Knölker, H.J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef]
- Poulsen, T.B.; Jørgensen, K.A. Catalytic asymmetric Friedel-Crafts alkylation reactions—Copper showed the way. Chem. Rev. 2008, 108, 2903–2915. [Google Scholar] [CrossRef]
- Nie, J.; Guo, H.C.; Cahard, D.; Ma, J.A. Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev. 2011, 111, 455–529. [Google Scholar] [CrossRef]
- Fuson, R.C.; Weinstock, H.H.; Ullyot, G.E. A new synthesis of benzoins. 2′,4′,6′-trimethylbenzoin. J. Am. Chem. Soc. 1935, 57, 1803–1804. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Sedaghati, M.; Schnakenburg, G. Reaction of indoles with aromatic fluoromethyl ketones: An efficient synthesis of trifluoromethyl(indolyl)phenylmethanols using K2CO3/n-Bu4PBr in water. Beilstein J. Org. Chem. 2020, 16, 778–7490. [Google Scholar] [CrossRef] [Green Version]
- Harikrishnan, A.; Selvakumar, J.; Gnanamani, E.; Bhattacharya, S.; Ramanathan, C.R. Friedel-Crafts hydroxyalkylation through activation of a carbonyl group using AlBr3: An easy access to pyridyl aryl/heteroaryl carbinols. New J. Chem. 2013, 37, 563–567. [Google Scholar] [CrossRef]
- Medimagh, R.; Marque, S.; Prim, D.; Chatti, S. A facile preparation of trisubstituted amino-furan and -thiophene derivatives. Org. Biomol. Chem. 2011, 9, 6055–6065. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Umemoto, T. Electrophilic perfluoroalkylating agents. Chem. Rev. 1996, 96, 1757–1777. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Yudin, A.K. Perfluoroalkylation with organosilicon reagents. Chem. Rev. 1997, 97, 757–786. [Google Scholar] [CrossRef]
- Kirij, N.V.; Yagupolskii, Y.L.; Petukh, N.V.; Tyrra, W.; Naumann, D. Trifluoromethylation of heterocumulenes with trimethyl(trifluoromethyl)silane in the presence of fluoride ions: Synthesis of trifluoroacetamides and trifluorothioacetamides from isocyanates and isothiocyanates. Tetrahedron Lett. 2001, 42, 8181–8183. [Google Scholar] [CrossRef]
- Horváth, I.T.; Anastas, P.T. Innovations and green chemistry. Chem. Rev. 2007, 107, 2169–2173. [Google Scholar] [CrossRef] [Green Version]
- Trost, B.M. On inventing reactions for atom economy. Acc. Chem. Res. 2002, 35, 695–705. [Google Scholar] [CrossRef]
- Trost, B.M. Atom economy—A challenge for organic synthesis: Homogeneous catalysis leads the way. Angew. Chem. Int. Ed. Engl. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2009, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Boibessot, T.; Zschiedrich, C.P.; Dunyach-Rémy, C.; Lebeau, A.; Bénimèlis, D.; Dunyach-Rémy, C.; Lavigne, J.-P.; Szurmant, H.; Benfodda, Z.; Meffre, P. The rational design, synthesis, and antimicrobial properties of thiophene derivatives that inhibit bacterial histidine kinases. J. Med. Chem. 2016, 59, 8830–8847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Gamage, T.F.; Decker, A.M.; Barrus, D.; Langston, T.L.; Li, J.-X.; Thomas, B.F.; Zhang, Y. Synthesis and pharmacological evaluation of 1-phenyl-3-thiophenylurea derivatives as cannabinoid type-1 receptor allosteric modulators. J. Med. Chem. 2019, 62, 9806–9823. [Google Scholar] [CrossRef] [PubMed]
- Garnett, J.L.; Sollich-Baumgartner, W.A. Catalytic deuterium exchange reactions with organics. XIV. Distinction between associative and dissociative π-complex substitution mechanisms. J. Phys. Chem. 1964, 68, 3177–3183. [Google Scholar] [CrossRef]
- Khodakovskiy, P.V.; Mykhailiuk, P.K.; Volochnyuk, D.M.; Tolmachev, A.A. Noncatalytic electrophilic oxyalkylation of some five-membered heterocycles with 2-(trifluoroacetyl)-1,3-azoles. Synthesis 2010, 979–984. [Google Scholar] [CrossRef]
- Nenajdenko, V.G.; Sanin, A.V.; Balenkova, E.S. Preparation of α,β-unsaturated ketones bearing a trifluoromethyl group and their application in organic synthesis. Molecules 1997, 2, 186–232. [Google Scholar] [CrossRef] [Green Version]
- Ho, T.L. The hard soft acids bases (HSAB) principle and organic chemistry. Chem. Rev. 1975, 75, 1–20. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Hedley, S.J. Intermolecular reactions of electron-rich heterocycles with copper and rhodium carbenoids. Chem. Soc. Rev. 2007, 36, 1109–1119. [Google Scholar] [CrossRef]
- Smith, M.B. March’s Advanced Organic Chemistry, 7th ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; ISBN 978-0-470-46259-1. [Google Scholar]
- Li, C.J. Organic reactions in aqueous media with a focus on carbon-carbon bond formations: A decade update. Chem. Rev. 2005, 105, 3095–3165. [Google Scholar] [CrossRef]
- Ding, R.; Zhang, H.B.; Chen, Y.J.; Liu, L.; Wang, D.; Li, C.J. In(OTf)3-catalyzed Friedel-Crafts reaction of aromatic compounds with methyl trifluoropyruvate in water. Synlett 2004, 3, 555–557. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
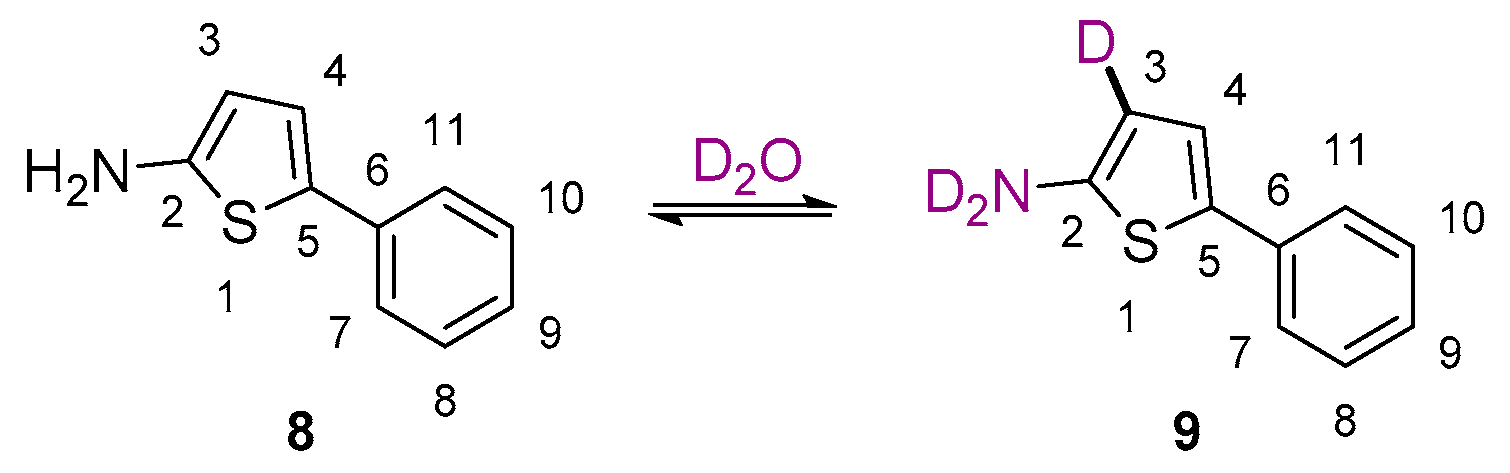{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | 11e Equivalent (eq.) | T (°C) | Yield (%) b |
|---|---|---|---|
| 1 a | 1 | 100 | 70% |
| 2 a | 1 | 120 | 83% |
| 3 a | 1 | 140 | 47% |
| 4 a | 1.5 | 120 | 82% |
| 5 a | 2 | 120 | 81% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duvauchelle, V.; Bénimélis, D.; Meffre, P.; Benfodda, Z. Catalyst-Free Site Selective Hydroxyalkylation of 5-Phenylthiophen-2-amine with α-Trifluoromethyl Ketones through Electrophilic Aromatic Substitution. Molecules 2022, 27, 925. https://doi.org/10.3390/molecules27030925
Duvauchelle V, Bénimélis D, Meffre P, Benfodda Z. Catalyst-Free Site Selective Hydroxyalkylation of 5-Phenylthiophen-2-amine with α-Trifluoromethyl Ketones through Electrophilic Aromatic Substitution. Molecules. 2022; 27(3):925. https://doi.org/10.3390/molecules27030925
Chicago/Turabian StyleDuvauchelle, Valentin, David Bénimélis, Patrick Meffre, and Zohra Benfodda. 2022. "Catalyst-Free Site Selective Hydroxyalkylation of 5-Phenylthiophen-2-amine with α-Trifluoromethyl Ketones through Electrophilic Aromatic Substitution" Molecules 27, no. 3: 925. https://doi.org/10.3390/molecules27030925