
Alpha-Glucosidase Inhibition and Molecular Docking of Isolated Compounds from Traditional Thai Medicinal Plant, Neuropeltis racemosa Wall.



Abstract
:
1. Introduction
2. Results
2.1. Determination of Alpha-Glucosidase Inhibition
2.2. Extraction, Isolation, and Identification of Pure Compounds
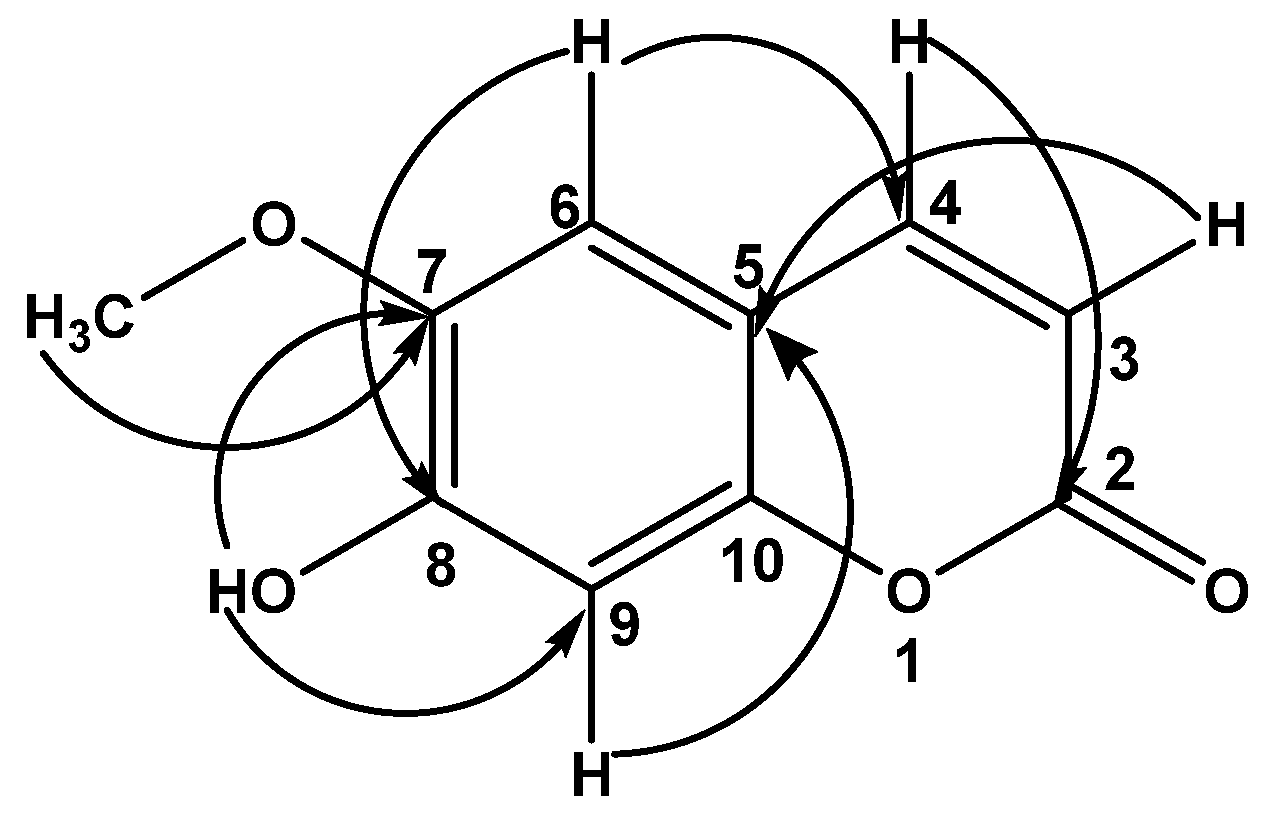
2.2.1. Scopoletin (1)
2.2.2. Syringic Acid (2)
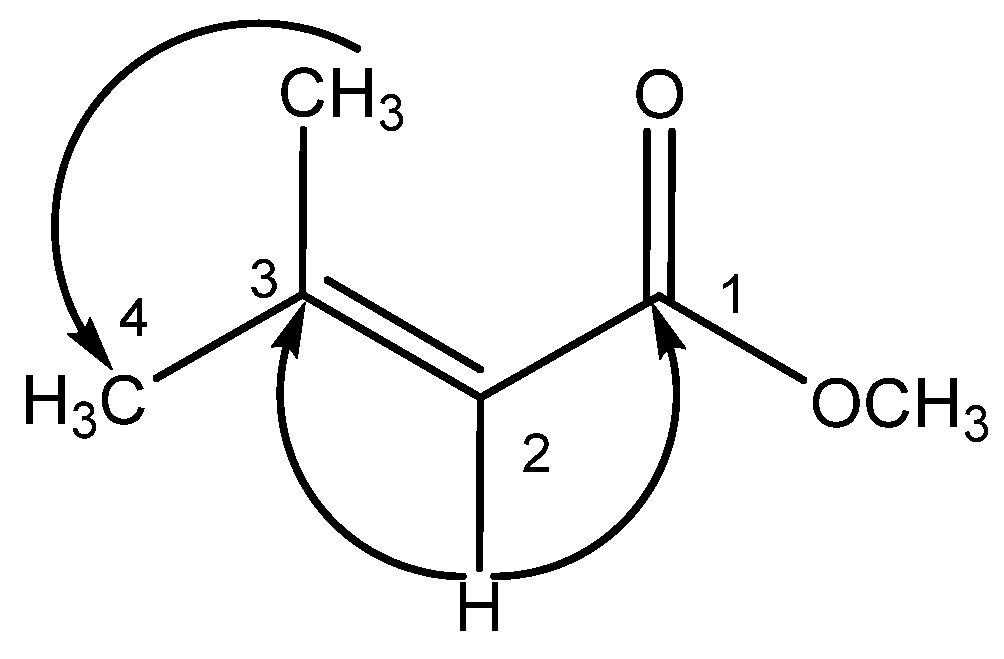
2.2.3. Methyl 3-Methyl-2-butenoate (3)
2.2.4. N-trans-Feruloyltyramine (4)
2.2.5. N-trans-Coumaroyltyramine (5)
2.3. Alpha-Glucosidase Inhibitory Assay
2.4. Enzyme Kinetic Assay
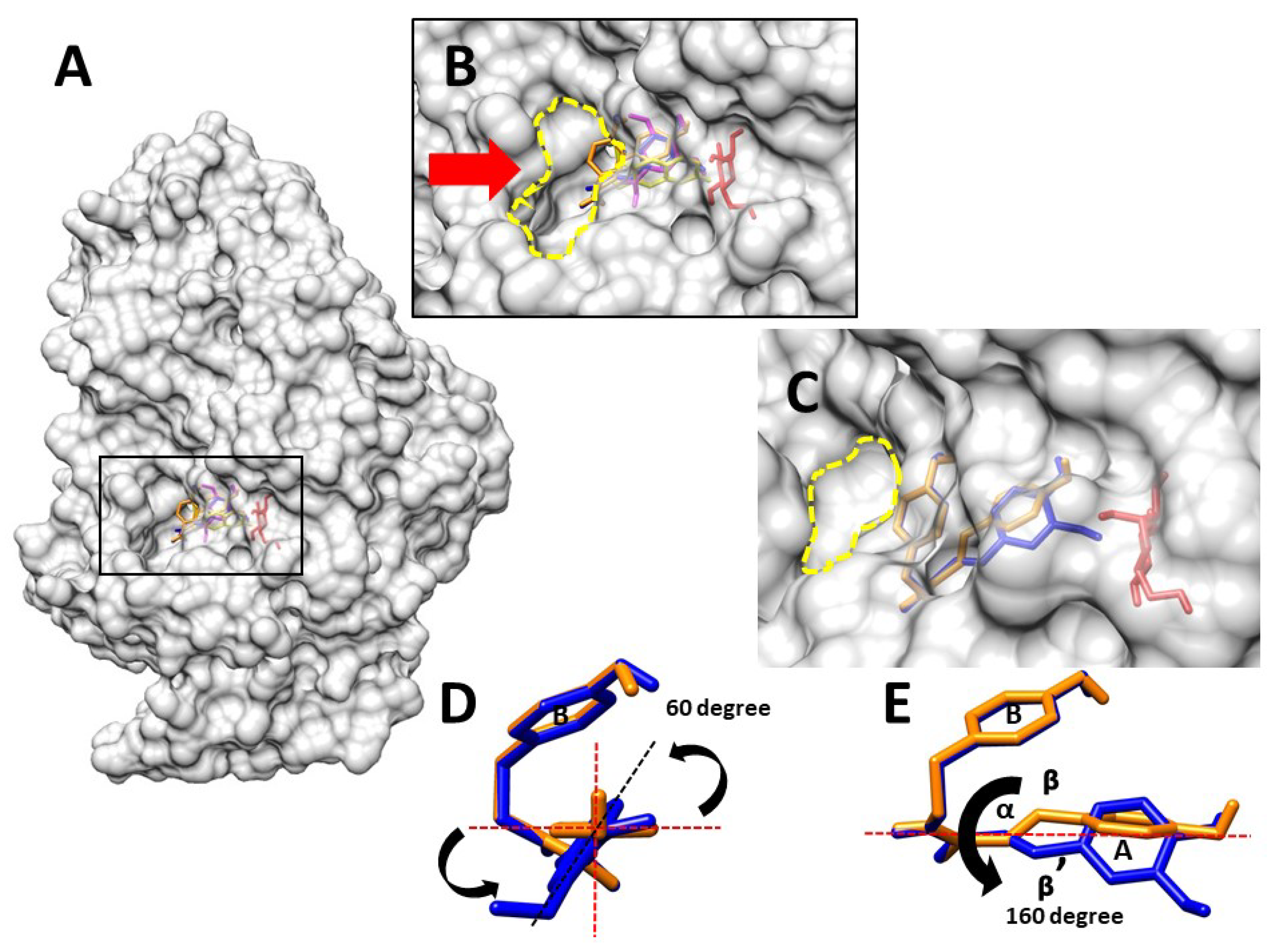
2.5. Molecular Docking
3. Discussion
4. Experimental Section
4.1. General
4.2. Plant Material
4.3. Extraction and Isolation
4.4. Spectroscopic Data
4.4.1. Scopoletin (1)
4.4.2. Syringic Acid (2)
4.4.3. Methyl 3-Methyl-2-butenoate (3)
4.4.4. N-trans-Feruloyltyramine (4)
4.4.5. N-trans-Coumaroyltyramine (5)
4.5. Enzymatic Assay
4.6. Enzyme Kinetic Study
4.7. Molecular Docking
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- IDF Diabetes Atlas Tenth Edition 2021. International Diabetes Federation (IDF). Available online: https://diabetesatlas.org/idfawp/resource-files/2021/07/IDF_Atlas_10th_Edition_2021.pdf (accessed on 25 December 2021).
- American Diabetes Association (ADA). Pharmacologic Approaches to Glycemic Treatment; American Diabetes Association: Arlington County, VA, USA, 2017; (Suppl. 1), pp. S64–S74. [Google Scholar]
- Chaudhury, A.; Duvoor, C.; Dendi, V.S.R.; Kraleti, S.; Chada, A.; Ravilla, R.; Marco, A.; Shekhawat, N.S.; Montales, M.T.; Kuriakose, K.; et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front. Endocrinol. 2017, 8, 6. [Google Scholar] [CrossRef] [Green Version]
- Santisuk, T.; Larsen, K. Flora of Thailand Volume 10 Part 3 Anacardiaceae & Convolvulaceae; Prachachod Co. Ltd.: Bangkok, Thailand, 2010; pp. 440–450. [Google Scholar]
- Art of Healing Division. General Ancient Medicine Textbook in Pharmaceutic; Ministry of Public Health: Bangkok, Thailand, 1998; p. 62. [Google Scholar]
- Chayarop, K.; Peungvicha, P.; Temsiririrkkul, R.; Wongkrajang, Y.; Chuakul, W.; Rojsanga, P. Hypoglycaemic activity of Mathurameha, a Thai traditional herbal formula aqueous extract, and its effect on biochemical profiles of streptozotocin-nicotinamide-induced diabetes rats. BMC Complement. Altern. Med. 2017, 17, 343. [Google Scholar] [CrossRef] [PubMed]
- Satean, G.; Homhuan, S.; Premaprasit, S. Plant utilization from Thungluilai community forest in Thungluilai sub-district, Kornsarn distric t, Chaiyaphum province. J. Soc. Sci. Naresuan Univ. 2018, 19, 211–245. [Google Scholar]
- Halabi, S.; Battah, A.A.; Aburjai, T.; Hudaib, M. Phytochemical and antiplatelet investigation of Gundelia tournifortii. Pharm. Biol. 2005, 43, 496–500. [Google Scholar] [CrossRef]
- Costa, T.M.; Mayer, D.A.; Siebert, D.A.; Micke, G.A.; Alberton, M.D.; Tavares, L.B.B.; Oliveira, D. Kinetics analysis of the inhibitory effects of alpha-glucosidase and identification of compounds from Ganoderma lipsiense mycelium. Appl. Biochem. Biotechnol. 2020, 191, 996–1009. [Google Scholar] [CrossRef]
- Mahomoodally, M.F.; Vlaisavljevic, S.; Berezni, S.; Abdallah, H.H.; Zengin, G.; Atanasov, A.G.; Mollica, A.; Lobine, D.; Aktumsek, A.; Abdurrahman, A. Lotus aegaeus (Gris.) Boiss and Iberis sempervirens L.: Chemical fingerprints, antioxidant potential, and inhibition activities and docking on key enzymes linked to global health problems. Ind. Crop Prod. 2018, 120, 271–278. [Google Scholar] [CrossRef]
- Song, Y.H.; Kim, D.W.; Curtis-Long, M.J.; Park, C.; Son, M.; Kim, J.Y.; Yuk, H.J.; Lee, K.W.; Park, K.H. Cinnamic acid amides from Tribulus terrestris displaying uncompetitive α-glucosidase inhibition. Eur. J. Med. Chem. 2016, 114, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, J.; Kong, L. Phenylethyl cinnamides as potential α-glucosidase inhibitors from the roots of Solanum melongena. Nat. Prod. Commun. 2011, 6, 851–853. [Google Scholar] [CrossRef] [Green Version]
- Panidthananon, W.; Chaowasku, T.; Sritularak, B.; Likhitwitayawuid, K. A new benzophenone C-glucoside and other constituents of Pseuduvaria fragrans and their α-glucosidase inhibitory activity. Molecules 2018, 23, 1600. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.N.; Jung, H.A.; Sohn, H.S.; Kim, H.M.; Choi, J.S. Potent α-glucosidase and protein tyrosine phosphatase 1Binhibitors from Artemisia capillaris. Arch. Pharm. Res. 2013, 36, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.C.; Ku, I.I.; Nur, K.K.; Lima, P.C.; Ismaila, I.S.; Hamidb, M.; Nga, R.C. Comparative study of the antidiabetic potential of Paederia foetida twig extracts and compounds from two different locations in Malaysia. Pharm. Biol. 2019, 57, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheemanapalli, S.; Mopuri, R.; Golla, R.; Anurudha, C.M.; Chitta, S.K. Syringic acid (SA)—A review of its occurrence, biosynthesis, pharmacological and industrial importance. Biomed. Pharmacother. 2018, 108, 547–557. [Google Scholar]
- Copeland, R.A. Reversible modes of inhibitor interactions with enzymes. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists; John Wiley & Sons: Hoboken, NJ, USA, 2005; pp. 48–81. [Google Scholar]
- Jang, J.H.; Park, J.E.; Han, J.S. Scopoletin inhibits α-glucosidase in vitro and alleviates postprandial hyperglycemia in mice with diabetes. Eur. J. Pharmacol. 2018, 834, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bai, B.; Liu, X.; Wang, Y.; Li, M.; Zhao, D. α-Glucosidase inhibitors from Chinese yam (Dioscorea opposite Thunb.). Food Chem. 2011, 126, 203–206. [Google Scholar] [CrossRef]
- Wu, Q.; Yang, X.; Zou, L.; Fu, D. Bioactivity guided isolation of alpha-glucosidase inhibitor from whole herbs of Crossostephium chinense. Zhongguo Zhong Yao Za Zhi 2009, 34, 2206–2211. [Google Scholar] [PubMed]
- Son, H.U.; Yoon, E.K.; Yoo, C.Y.; Park, C.H.; Bae, M.A.; Kim, T.H.; Lee, C.H.; Lee, K.W.; Seo, H.; Kim, K.J.; et al. Effects of synergistic inhibition on α-glucosidase by phytoalexins in soybeans. Biomolecules 2019, 9, 828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dej-adisai, S.; Pitakbut, T. Determination of α-glucosidase inhibitory activity from selected Fabaceae plants. Pak. J. Pharm. Sci. 2015, 28, 1679–1683. [Google Scholar]
- Chetty, S.; Soliman, M.E.S. Possible allosteric binding site on Gyrase B, a key target for novel anti-TB drugs: Homology modelling and binding site identification using molecular dynamics simulation and binding free energy calculations. Med. Chem. Res. 2015, 24, 2055–2074. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dej-adisai, S.; Rais, I.R.; Wattanapiromsakul, C.; Pitakbut, T. Alpha-glucosidase inhibitory assay-screened isolation and molecular docking model from Bauhinia Pulla active compounds. Molecules 2021, 26, 5970. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µg/mL) | Inhibition Type | Ki b or Ki′ c (µM) |
|---|---|---|---|
| Scopoletin (1) | 110.97 | Mixed | - |
| Syringic acid (2) | >500 | N.T. a | - |
| Methyl 3-methyl-2-butenoate (3) | >500 | N.T. a | - |
| N-trans-feruloyltyramine (4) | 29.87 | Uncompetitive | 51.81 c |
| N-trans-coumaroyltyramine (5) | 0.92 | Uncompetitive | 1.99 c |
| Acarbose (Positive control) | 272.72 | Competitive | 264.46 b |
| Complex | Number(s) of Interaction | Interaction Sites | Distances (Å) |
|---|---|---|---|
| AR—1 | 1 | Glucose 601 | 1—Glucose 601 (3.41 Å) |
| AR—2 | 4 | Tyr 158 | 2—Tyr 158 (2.05 Å) |
| Glucose 601 | 2—Glucose 601 (1.36 Å) | ||
| 2—Glucose 601 (3.14 Å) | |||
| 2—Glucose 601 (4.21 Å) | |||
| AR—4 | 4 | Arg 315 | 4—Arg 315 (4.60 Å) |
| Asn 415 | 4—Asn 415 (4.18 Å) | ||
| Glucose 601 | 4—Glucose 601 (4.33 Å) | ||
| 4—Glucose 601 (3.21 Å) | |||
| AR—5 | 4 | Arg 315 | 5—Arg 315 (4.64 Å) |
| Asn 415 | 5—Asn 415 (4.28 Å) | ||
| Glucose 601 | 5—Glucose 601 (4.14 Å) | ||
| 5—Glucose 601 (2.88 Å) |
| Autodock 4.2.6 | Autodock Vina | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | vdW+Hbond (I) | Elec. Energy (II) | Desol. energy (III) | Total Intermol. Interact. Energy (IV; I+II+III) | Total Internal Energy (V) | Tors. Free Energy (VI) | Unbound’s Energy (VII) | Binding Energy (Kcal/mol) (VIII; IV+V+VI+VII) | Affinity (Kcal/mol) |
| 1 | −6.34 | 0.01 | 1.80 | −4.53 | −0.69 | 0.6 | 0 | −4.62 | −6.3 |
| 2 | −6.67 | 1.70 | 3.00 | −1.98 | −0.58 | 1.49 | 0 | −1.07 | −5.7 |
| 4 | −9.27 | 0.30 | 2.60 | −6.36 | −1.45 | 2.39 | 0 | −5.42 | −7.5 |
| 5 | −8.22 | 0.29 | 2.05 | −5.88 | −1.36 | 2.09 | 0 | −5.15 | −7.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sakulkeo, O.; Wattanapiromsakul, C.; Pitakbut, T.; Dej-adisai, S. Alpha-Glucosidase Inhibition and Molecular Docking of Isolated Compounds from Traditional Thai Medicinal Plant, Neuropeltis racemosa Wall. Molecules 2022, 27, 639. https://doi.org/10.3390/molecules27030639
Sakulkeo O, Wattanapiromsakul C, Pitakbut T, Dej-adisai S. Alpha-Glucosidase Inhibition and Molecular Docking of Isolated Compounds from Traditional Thai Medicinal Plant, Neuropeltis racemosa Wall. Molecules. 2022; 27(3):639. https://doi.org/10.3390/molecules27030639
Chicago/Turabian StyleSakulkeo, Oraphan, Chatchai Wattanapiromsakul, Thanet Pitakbut, and Sukanya Dej-adisai. 2022. "Alpha-Glucosidase Inhibition and Molecular Docking of Isolated Compounds from Traditional Thai Medicinal Plant, Neuropeltis racemosa Wall." Molecules 27, no. 3: 639. https://doi.org/10.3390/molecules27030639