
Metabolite Profiling and Bioassay-Guided Fractionation of Zataria multiflora Boiss. Hydroethanolic Leaf Extracts for Identification of Broad-Spectrum Pre and Postharvest Antifungal Agents

Abstract
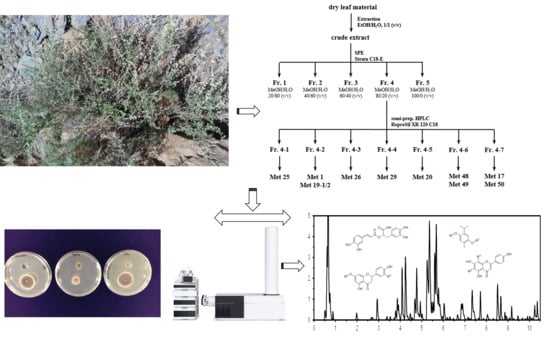
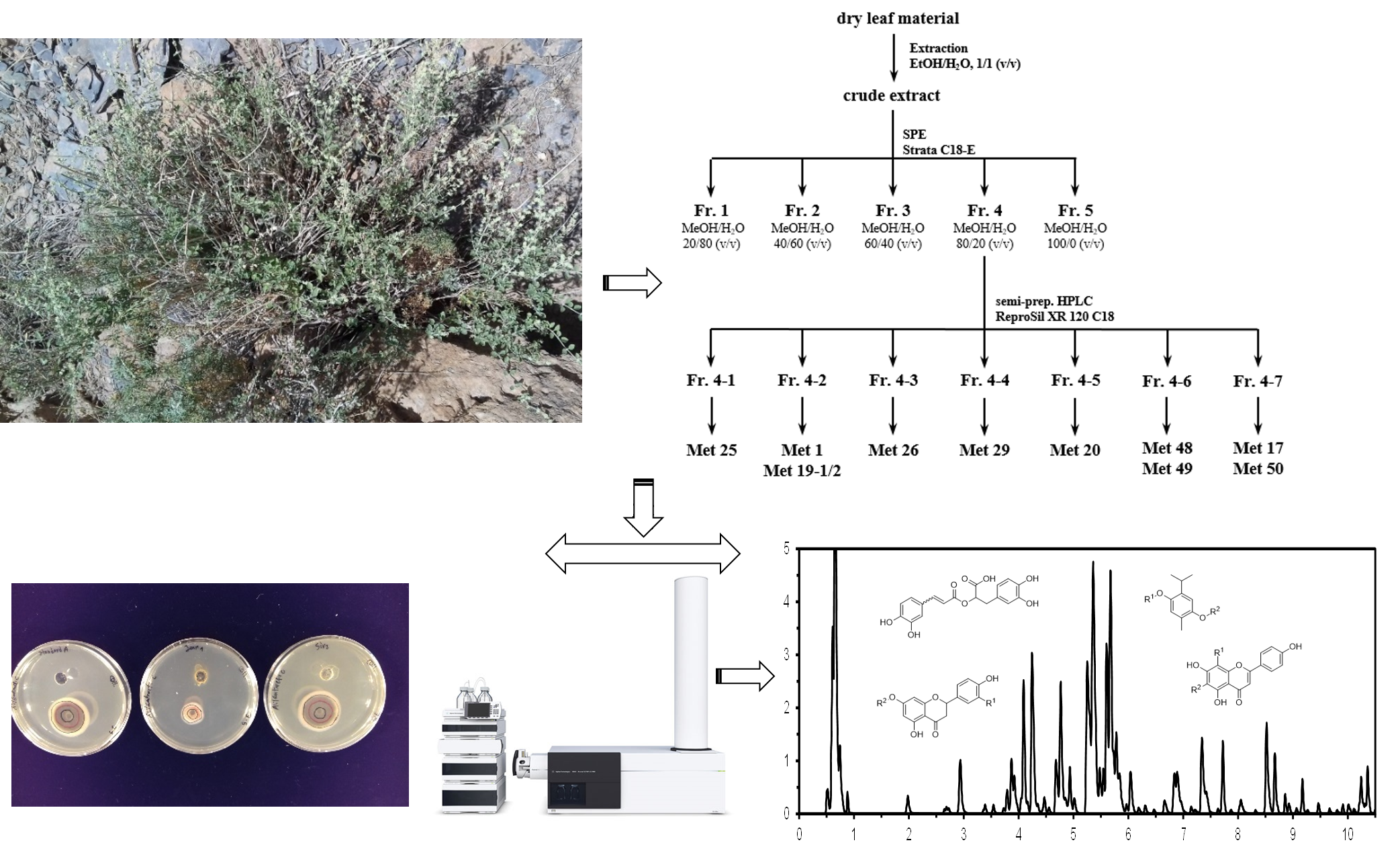
:
1. Introduction
2. Results
2.1. Screening of Antifungal Activities of Hydroethanolic Leaf Extracts
2.2. Metabolite Profiling of Hydroethanolic Leaf Extracts
2.3. Annotation of Semi-Polar Secondary Metabolites
2.3.1. Flavonoids
2.3.2. Hydroxycinnamic Acid Derivatives
2.3.3. Terpenoids
2.3.4. Other Metabolites
2.4. Correlation of Metabolite Abundance with Antifungal Activity
2.5. Bioassay-Guided Fractionation of a Hydroethanolic Leaf Extract
3. Discussion
4. Experimental
4.1. Plant Material, Fungal Species and Chemicals
4.2. Preparation of Hydroethanolic Leaf Extracts for Bioassays and Metabolite Analyses
4.3. Antifungal Assays
4.4. Fractionation of Hydroethanolic Leaf Extracts
4.5. UHPLC/DAD/ESI-QTOFMS
4.6. LC/MS Data Processing
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Frezza, C.; Venditti, A.; Serafini, M.; Bianco, A. Phytochemistry, chemotaxonomy, ethnopharmacology, and nutraceutics of Lamiaceae. Stud. Nat. Prod. Chem. 2019, 62, 125–178. [Google Scholar]
- Sajed, H.; Sahebkar, A.; Iranshahi, M. Zataria multiflora Boiss. (Shirazi thyme)—An ancient condiment with modern pharmaceutical uses. J. Ethnopharmacol. 2013, 145, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Krähmer, A.; Herwig, N.; Schulz, H.; Hadian, J.; Meiners, T. Variation of secondary metabolite profile of Zataria multiflora Boiss. populations linked to geographic, climatic, and edaphic factors. Front. Plant Sci. 2020, 11, 969. [Google Scholar] [CrossRef] [PubMed]
- Khazdair, M.R.; Ghorani, V.; Alavinezhad, A.; Boskabady, M.H. Effect of Zataria multiflora on serum cytokine levels and pulmonary function tests in sulfur mustard-induced lung disorders: A randomized double-blind clinical trial. J. Ethnopharmacol. 2020, 248, 112325. [Google Scholar] [CrossRef]
- Izadiyan, P.; Hemmateenejad, B.; Francisco, M.; Rodríguez, V.M.; Izadiyan, M. Metabolite fingerprinting and identification of potential quality markers of Zataria multiflora by a chemometric approach. J. Iran. Chem. Soc. 2019, 16, 1631–1639. [Google Scholar] [CrossRef]
- Ali, M.S.; Saleem, M.; Akhtar, F.; Jahangir, M.; Parvez, M.; Ahmad, V.U. Three p-cymene derivatives from Zataria multiflora. Phytochemistry 1999, 52, 685–688. [Google Scholar] [CrossRef]
- Ali, M.S.; Saleem, M.; Ali, Z.; Ahmad, V.U. Chemistry of Zataria multiflora (lamiaceae). Phytochemistry 2000, 55, 933–936. [Google Scholar]
- Bebber, D.P.; Gurr, S.J. Crop-destroying fungal and oomycete pathogens challenge food security. Fungal Genet. Biol. 2015, 74, 62–64. [Google Scholar] [CrossRef]
- Fraeyman, S.; Croubels, S.; Devreese, M.; Antonissen, G. Emerging Fusarium and Alternaria mycotoxins: Occurrence, toxicity and toxicokinetics. Toxins 2017, 9, 228. [Google Scholar] [CrossRef] [Green Version]
- Williamson, B.; Tudzynski, B.; Tudzynski, P.; Van Kan, J.A. Botrytis cinerea: The cause of grey mould disease. Mol. Plant Pathol. 2007, 8, 561–580. [Google Scholar] [CrossRef]
- Shuping, D.S.S.; Eloff, J.N. The use of plants to protect plants and food against fungal pathogens: A review. Afr. J. Tradit. Complement. Altern. Med. 2017, 14, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayan, F.E.; Cantrell, C.L.; Duke, S.O. Natural products in crop protection. Bioorg. Med. Chem. 2009, 17, 4022–4034. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.; Meiners, T. Antifungal activity of Zataria multiflora Boiss. essential oils and changes in volatile compound composition under abiotic stress conditions. Ind. Crops Prod. 2021, 171, 113888. [Google Scholar] [CrossRef]
- Wang, Z.L.; Gao, H.M.; Wang, S.; Zhang, M.; Chen, K.; Zhang, Y.Q.; Wang, H.D.; Han, B.Y.; Xu, L.L.; Song, T.Q.; et al. Dissection of the general two-step di-C-glycosylation pathway for the biosynthesis of (iso) schaftosides in higher plants. Proc. Natl. Acad. Sci. USA 2020, 117, 30816–30823. [Google Scholar] [CrossRef] [PubMed]
- Grayer, R.J.; Eckert, M.R.; Veitch, N.C.; Kite, G.C.; Marin, P.D.; Kokubun, T.; Simmonds, M.S.; Paton, A.J. The chemotaxonomic significance of two bioactive caffeic acid esters, nepetoidins A and B, in the Lamiaceae. Phytochemistry 2003, 64, 519–528. [Google Scholar] [CrossRef]
- Böttcher, C.; Chapman, A.; Fellermeier, F.; Choudhary, M.; Scheel, D.; Glawischnig, E. The biosynthetic pathway of indole-3-carbaldehyde and indole-3-carboxylic acid derivatives in Arabidopsis. Plant Physiol. 2014, 165, 841–853. [Google Scholar] [CrossRef] [Green Version]
- Saldanha, L.L.; Allard, P.M.; Afzan, A.; de Melo, F.P.D.S.R.; Marcourt, L.; Queiroz, E.F.; Vilegas, W.; Furlan, C.M.; Dokkedal, A.L.; Wolfender, J.L. Metabolomics of Myrcia bella populations in Brazilian Savanna reveals strong influence of environmental factors on its specialized metabolism. Molecules 2020, 25, 2954. [Google Scholar] [CrossRef]
- Brunetti, C.; Fini, A.; Sebastiani, F.; Gori, A.; Tattini, M. Modulation of phytohormone signaling: A primary function of flavonoids in plant–environment interactions. Front. Plant Sci. 2018, 9, 1042. [Google Scholar] [CrossRef] [Green Version]
- Wolfender, J.L.; Litaudon, M.; Touboul, D.; Queiroz, E.F. Innovative omics-based approaches for prioritisation and targeted isolation of natural products—New strategies for drug discovery. Nat. Prod. Rep. 2019, 36, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Martins, N.; Barros, L.; Santos-Buelga, C.; Silva, S.; Henriques, M.; Ferreira, I.C. Decoction, infusion and hydroalcoholic extract of cultivated thyme: Antioxidant and antibacterial activities, and phenolic characterisation. Food Chem. 2015, 167, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Jordán, M.J.; Lax, V.; Rota, M.C.; Lorán, S.; Sotomayor, J.A. Relevance of carnosic acid, carnosol, and rosmarinic acid concentrations in the in vitro antioxidant and antimicrobial activities of Rosmarinus officinalis (L.) methanolic extracts. J. Agric. Food. Chem. 2012, 60, 9603–9608. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.B.; Rai, D.K.; Brunton, N.P.; Martin-Diana, A.B.; Barry-Ryan, C. Characterization of phenolic composition in Lamiaceae spices by LC-ESI-MS/MS. J. Agric. Food. Chem. 2010, 58, 10576–10581. [Google Scholar] [CrossRef] [PubMed]
- Funari, C.S.; Gullo, F.P.; Napolitano, A.; Carneiro, R.L.; Mendes-Giannini, M.J.S.; Fusco-Almeida, A.M.; Piacente, S.; Pizza, C.; Silva, D.H.S. Chemical and antifungal investigations of six Lippia species (Verbenaceae) from Brazil. Food Chem. 2012, 135, 2086–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Förster, C.; Handrick, V.; Ding, Y.; Nakamura, Y.; Paetz, C.; Schneider, B.; Castro-Falcón, G.; Hughes, C.C.; Luck, K.; Poosapati, S.; et al. Biosynthesis and antifungal activity of fungus-induced O-methylated flavonoids in maize. Plant Physiol. 2022, 188, 167–190. [Google Scholar] [CrossRef]
- Kodama, O.; Miyakawa, J.; Akatsuka, T.; Kiyosawa, S. Sakuranetin, a flavanone phytoalexin from ultraviolet-irradiated rice leaves. Phytochemistry 1992, 31, 3807–3809. [Google Scholar] [CrossRef]
- Vergine, M.; Nicolì, F.; Negro, C.; Luvisi, A.; Nutricati, E.; Accogli, R.A.; Sabella, E.; Miceli, A. Phytochemical profiles and antioxidant activity of Salvia species from southern Italy. Rec. Nat. Prod. 2019, 13, 215. [Google Scholar] [CrossRef]
- Koukoulitsa, C.; Karioti, A.; Bergonzi, M.C.; Pescitelli, G.; Di Bari, L.; Skaltsa, H. Polar constituents from the aerial parts of Origanum vulgare L. ssp. hirtum growing wild in Greece. J. Agric. Food. Chem. 2006, 54, 5388–5392. [Google Scholar] [CrossRef]
- Barros, L.; Dueñas, M.; Dias, M.I.; Sousa, M.J.; Santos-Buelga, C.; Ferreira, I.C. Phenolic profiles of cultivated, in vitro cultured and commercial samples of Melissa officinalis L. infusions. Food Chem. 2013, 136, 1–8. [Google Scholar] [CrossRef]
- Ly, T.N.; Shimoyamada, M.; Yamauchi, R. Isolation and characterization of rosmarinic acid oligomers in Celastrus hindsii Benth leaves and their antioxidative activity. J. Agric. Food. Chem. 2006, 54, 3786–3793. [Google Scholar] [CrossRef]
- Krause, S.T.; Liao, P.; Crocoll, C.; Boachon, B.; Förster, C.; Leidecker, F.; Wiese, N.; Zhao, D.; Wood, J.C.; Buell, C.R.; et al. The biosynthesis of thymol, carvacrol, and thymohydroquinone in Lamiaceae proceeds via cytochrome P450s and a short-chain dehydrogenase. Proc. Natl. Acad. Sci. USA 2021, 118, e2110092118. [Google Scholar] [CrossRef]
- Exarchou, V.; Kanetis, L.; Charalambous, Z.; Apers, S.; Pieters, L.; Gekas, V.; Goulas, V. HPLC-SPE-NMR characterization of major metabolites in Salvia fruticosa Mill. extract with antifungal potential: Relevance of carnosic acid, carnosol, and hispidulin. J. Agric. Food. Chem. 2015, 63, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Park, J.R.; Kim, K.M. Antimicrobial activity of chrysoeriol 7 and chochlioquinone 9, white-backed Planthopper-resistant compounds, against rice pathogenic strains. Biology 2020, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Al Aboody, M.S.; Mickymaray, S. Anti-fungal efficacy and mechanisms of flavonoids. Antibiotics 2020, 9, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, A.K.; Shah, M.R.; Anis, I.; Marwat, I.K. In vitro antifungal and antibacterial activities of extracts of Galium tricornutum subsp. longipedunculatum. J. Enzyme Inhib. Med. Chem. 2009, 24, 192–196. [Google Scholar] [CrossRef]
- Sen, A.; Batra, A. Evaluation of antimicrobial activity of different solvent extracts of medicinal plant: Melia azedarach L. Int. J. Curr. Pharm. Res. 2012, 4, 67–73. [Google Scholar]
- Tais, L.; Schulz, H.; Böttcher, C. Comprehensive profiling of semi-polar phytochemicals in whole wheat grains (Triticum aestivum) using liquid chromatography coupled with electrospray ionization quadrupole time-of-flight mass spectrometry. Metabolomics 2021, 17, 18. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Code | Inhibition of Mycelial Growth (%) | ||||
|---|---|---|---|---|---|---|
| F. culmorum | F. sambucinum | B. cinerea | A. dauci | C. lindemuthianum | ||
| Arsenjan | Ars | 51.5 ± 2.5 a,b,c | 50.3 ± 3.8 a,b,c | 50.5 ± 1.5 a,b,c | 58.7 ± 3.1 b | 48.2 ± 5.5 d,e |
| Ashkezar | Ash | 49.3 ± 3.1 b,c | 40.5 ± 3.3 d | 46.1 ± 1.4 b,c,d | 55.1 ± 1.5 b,c | 44.8 ± 2.8 e |
| Daarbast | Daa | 57.5 ± 2.5 a | 51.4 ± 3.6 a,b | 53.9 ± 3.7 a | 58.1 ± 3.9 b | 65.7 ± 4.2 a |
| Darab | Dar | 52.2 ± 2.9 a,b,c | 46.4 ± 2.3 b,c,d | 44.2 ± 2.3 c,d,e | 51.6 ± 1.1 c,d | 47.6 ± 2.7 e |
| Fasa | Fas | 52.9 ± 2.8 a,b,c | 43.2 ± 1.5 b,c,d | 40.1 ± 7.7 d,e | 48.4 ± 1.7 d | 36.1 ± 3.7 f |
| Gachooyeh | Gac | 54.6 ± 0.7 a,b,c | 47.6 ± 7.4 b,c,d | 46.9 ± 1.6 a,b,c,d | 53.5 ± 1.3 b,c,d | 46.2 ± 2.2 e |
| Gezeh | Gez | 56.2 ± 3.4 a,b | 48.4 ± 2.7 a,b,c,d | 50.9 ± 1.5 a,b,c | 58.3 ± 4.7 b | 55.4 ± 2.7 c |
| Haneshk | Han | 36.8 ± 2.9 d | 24.4 ± 10.5 e | 21.7 ± 5.4 f | 36.7 ± 2.1 e | 22.5 ± 2.3 g |
| Hongooyeh | Hon | 54.5 ± 3.3 a,b,c | 57.6 ± 2.6 a | 51.2 ± 2.3 a,b,c | 56.4 ± 3.5 b,c | 48.7 ± 3.7 d,e |
| Jandaq | Jan | 57.3 ± 3.4 a | 50.9 ± 2.3 a,b,c | 54.4 ± 0.8 a | 64.9 ± 3.5 a | 62.6 ± 3.3 a,b |
| Kemeshk | Kem | 56.6 ± 8.9 a,b | 43.3 ± 6.4 b,c,d | 50.9 ± 4.2 a,b,c | 53.6 ± 3.4 b,c,d | 53.8 ± 4.1 c,d |
| Konar Siah | Kon | 59.1 ± 1.9 a | 51.7 ± 3.7 a,b | 51.9 ± 1.7 a,b | 59.6 ± 3.3 a,b | 59.1 ± 0.6 b,c |
| Siriz | Sir | 31.4 ± 3.6 d | 29.6 ± 3.8 e | 18.9 ± 8.3 f | 32.7 ± 4.8 e | 21.1 ± 3.6 g |
| Taft | Taf | 47.9 ± 3.8 c | 41.3 ± 4.5 c,d | 37.9 ± 0.5 e | 54.1 ± 3.4 b–d | 36.1 ± 4.2 f |
| No. | Compound | Elemental Composition | AL a | RT [min] | Quantifier Ion | ||
|---|---|---|---|---|---|---|---|
| Type | m/z | m/z | |||||
| Measured | Calculated | ||||||
| 1 | Apigenin | C15H10O5 | 1 | 7.34 | [M + H]+ | 271.0606 | 271.0601 |
| 2 | Apigenin C-Hex—isomer#1 | C21H20O10 | 2 | 5.01 | [M + H]+ | 433.1128 | 433.1129 |
| 3 | Apigenin C-Hex—isomer#2 | C21H20O10 | 2 | 5.25 | [M + H]+ | 433.1131 | 433.1129 |
| 4 | Apigenin 6-C-Hex-8-C-Hex | C27H30O15 | 2 | 4.23 | [M + H]+ | 595.1656 | 595.1657 |
| 5 | Apigenin 7-O-β-d-Glc | C21H20O10 | 1 | 5.75 | [M + H]+ | 433.1125 | 433.1129 |
| 6 | Apigenin O-HexA | C21H18O11 | 2 | 5.72 | [M + H]+ | 447.0923 | 447.0922 |
| 7 | Apigenin O-(O-DeoxyHex-Hex) | C27H30O14 | 2 | 5.65 | [M + H]+ | 579.1699 | 579.1708 |
| 8 | Luteolin | C15H10O6 | 1 | 6.82 | [M + H]+ | 287.0552 | 287.0550 |
| 9 | Luteolin C-Hex—isomer#1 | C21H20O11 | 2 | 4.69 | [M + H]+ | 449.1073 | 449.1078 |
| 10 | Luteolin C-Hex—isomer#2 | C21H20O11 | 2 | 4.76 | [M + H]+ | 449.1075 | 449.1078 |
| 11 | Luteolin 6-C-Hex-8-C-Hex | C27H30O16 | 2 | 3.92 | [M + H]+ | 611.1606 | 611.1607 |
| 12 | Luteolin 7-O-β-d-Glc | C21H20O11 | 1 | 5.27 | [M + H]+ | 449.1079 | 449.1078 |
| 13 | Luteolin O-HexA | C21H18O12 | 2 | 5.23 | [M + H]+ | 463.0871 | 463.0871 |
| 14 | Luteolin O-(O-DeoxyHex-Hex) | C27H30O15 | 2 | 5.25 | [M + H]+ | 595.1657 | 595.1657 |
| 15 | 6-Hydroxyluteolin O-Hex | C21H20O12 | 2 | 4.84 | [M − H]− | 463.088 | 463.0882 |
| 16 | 6-Hydroxyluteolin O-HexA | C21H18O13 | 2 | 4.8 | [M + H]+ | 479.0818 | 479.082 |
| 17 | Genkwanin | C16H12O5 | 1 | 8.55 | [M − H]− | 283.0612 | 283.0612 |
| 18 | Trihydroxy-methoxy-flavone | C16H12O6 | 3 | 6.8 | [M − H]− | 299.0561 | 299.0561 |
| 19-1 b | Hispidulin | C16H12O6 | 1 | 7.41 | [M − H]− | 299.0561 | 299.0561 |
| 19-2 b | Chrysoeriol | ||||||
| 20 | 7-O-Methylluteolin | C16H12O6 | 1 | 8.06 | [M − H]− | 299.0561 | 299.0561 |
| 21 | Chrysoeriol O-HexA | C22H20O12 | 2 | 5.83 | [M − H]− | 475.0883 | 475.0882 |
| 22 | Hispidulin O-Hex | C22H22O11 | 2 | 5.97 | [M − H]− | 461.1087 | 461.1089 |
| 23 | Dihydroxy-dimethoxy-flavone | C17H14O6 | 3 | 7.99 | [M + H]+ | 315.0862 | 315.0863 |
| 24 | Velutin | C17H14O6 | 2 | 8.54 | [M − H]− | 313.0712 | 313.0718 |
| 25 | Trihydroxy-dimethoxy-flavone | C17H14O7 | 3 | 6.87 | [M + H]+ | 331.0815 | 331.0812 |
| 26 | Dihydroxy-trimethoxy-flavone | C18H16O7 | 3 | 7.46 | [M + H]+ | 345.0969 | 345.0969 |
| 27 | Naringenin | C15H12O5 | 1 | 6.66 | [M + H]+ | 273.0762 | 273.0757 |
| 28 | Eriodictyol | C15H12O6 | 1 | 6.03 | [M + H]+ | 289.0707 | 289.0707 |
| 29 | Sakuranetin | C16H14O5 | 2 | 7.93 | [M + H]+ | 287.0913 | 287.0914 |
| 30 | 2-Hydroxynaringenin 6-C-Hex-8-C-Hex | C27H32O16 | 2 | 3.47 | [M − H]− | 611.1604 | 611.1618 |
| 31 | Dihydrokaempferol | C15H12O6 | 1 | 5.34 | [M + H]+ | 289.0709 | 289.0707 |
| 32 | Dihydroquercetin | C15H12O7 | 1 | 4.66 | [M − H]− | 303.0511 | 303.051 |
| 33 | Rosmarinic Acid (RA) | C18H16O8 | 1 | 5.69 | [M − H]− | 359.0773 | 359.0772 |
| 34 | RA Dehydrodimer | C36H30O16 | 3 | 5.6 | [M − H]− | 717.1461 | 717.1461 |
| 35 | RA Dehydrotrimer—isomer#1 | C54H44O24 | 3 | 5.48 | [M − H]− | 1075.2156 | 1075.215 |
| 36 | RA Dehydrotrimer—isomer#2 | C54H44O24 | 3 | 5.78 | [M − H]− | 1075.2149 | 1075.215 |
| 37 | RA Dehydrotetramer | C72H58O32 | 3 | 5.85 | [M − 2H]2− | 716.1388 | 716.1383 |
| 38 | Caffeic acid | C9H8O4 | 1 | 3.73 | [M − H]− | 179.0349 | 179.0350 |
| 39 | Chlorogenic acid | C16H18O9 | 1 | 3.36 | [M − H]− | 353.0873 | 353.0878 |
| 40 | Nepetoidin | C17H14O6 | 2 | 7.15 | [M − H]− | 313.0715 | 313.0718 |
| 41 | Coumaric acid 4-O-Hex | C15H18O8 | 2 | 3.47 | [M − H]− | 325.0916 | 325.0929 |
| 42 | Thymohydroquinone (THQ) | C10H14O2 | 2 | 7.73 | [M − H]− | 165.0923 | 165.0921 |
| 43 | THQ O-Hex—isomer#1 (Zataroside) | C16H24O7 | 2 | 3.79 | [M + HCOO]− | 373.1504 | 373.1504 |
| 44 | THQ O-Hex—isomer#2 (Zataroside) | C16H24O7 | 2 | 4.04 | [M + HCOO]− | 373.1499 | 373.1504 |
| 45 | THQ O-(O-Malonyl-Hex)—isomer#1 | C19H26O10 | 2 | 4.77 | [M − H − CO2]− | 369.1553 | 369.1555 |
| 46 | THQ O-(O-Malonyl-Hex)—isomer#2 | C19H26O10 | 2 | 4.93 | [M − H − CO2]− | 369.1552 | 369.1555 |
| 47 | THQ O-(O-Hex-Hex) | C22H34O12 | 2 | 2.65 | [M + HCOO]− | 535.2027 | 535.2032 |
| 48 | unknown diterpenoid#1 | C20H26O4 | 4 | 8.52 | [M − H]− | 329.1756 | 329.1758 |
| 49 | unknown diterpenoid#2 | C20H24O4 | 4 | 9.18 | [M − H]− | 327.1600 | 327.1602 |
| 50 | unknown diterpenoid#3 | C20H26O3 | 4 | 8.68 | [M − H]− | 313.1808 | 313.1809 |
| 51 | unknown diterpenoid#4 | C20H26O3 | 4 | 8.86 | [M − H]− | 313.1808 | 313.1809 |
| 52 | unknown diterpenoid#5 | C20H26O3 | 4 | 10.37 | [M − H]− | 313.1808 | 313.1809 |
| 53 | unknown diterpenoid#6 | C20H24O3 | 4 | 10.63 | [M − H]− | 311.1653 | 311.1653 |
| 54 | unknown diterpenoid#7 | C20H24O5 | 4 | 9.91 | [M − H]− | 343.1548 | 343.1551 |
| 55 | unknown diterpenoid#8 | C20H24O5 | 4 | 10.02 | [M − H]− | 343.1548 | 343.1551 |
| 56 | β-Glucosyl indole-3-carboxylate | C15H17NO7 | 2 | 3.87 | [M − H]− | 322.0932 | 322.0932 |
| 57 | Hydroxyjasmonic acid (Hydroxy-JA) | C12H18O4 | 3 | 4.45 | [M − H]− | 225.1132 | 225.1132 |
| 58 | Hydroxy-JA O-Hex | C18H28O9 | 3 | 4.08 | [M − H]− | 387.1659 | 387.1661 |
| 59 | Hydroxy-JA O-(O-Malonyl-Hex) | C21H30O12 | 3 | 4.64 | [M − H − CO2]− | 429.1759 | 429.1766 |
| 60 | 3,4-Dihydroxybenzaldehyde | C7H6O3 | 1 | 2.94 | [M − H]− | 137.0247 | 137.0244 |
| 61 | Dihydroxybenzoic acid O-Hex-O-Pent | C18H24O13 | 3 | 2.73 | [M − H]− | 447.1141 | 447.1144 |
| 62 | Dimethoxyhydroxybenzoic acid O-Hex | C15H20O10 | 3 | 2.94 | [M − H]− | 359.0981 | 359.0984 |
| 63 | Tyramine | C8H11NO | 1 | 0.88 | [M + H − NH3]+ | 121.0643 | 121.0648 |
| 64 | N-(γ-Glutamyl)-tyramine | C13H18N2O4 | 2 | 2.43 | [M + H]+ | 267.1337 | 267.1339 |
| 65 | Tyrosol 8-O-Hex | C14H20O7 | 2 | 2.98 | [M + NH4]+ | 318.1543 | 318.1547 |
| 66 | Tyrosol O-(O-Pent-Hex) | C19H28O11 | 2 | 3.39 | [M − H]− | 431.1556 | 431.1559 |
| 67 | Benzylalcohol O-(O-Malonyl-Hex) | C16H20O9 | 2 | 4.74 | [M + NH4]+ | 374.1440 | 374.1446 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karimi, A.; Meiners, T.; Böttcher, C. Metabolite Profiling and Bioassay-Guided Fractionation of Zataria multiflora Boiss. Hydroethanolic Leaf Extracts for Identification of Broad-Spectrum Pre and Postharvest Antifungal Agents. Molecules 2022, 27, 8903. https://doi.org/10.3390/molecules27248903
Karimi A, Meiners T, Böttcher C. Metabolite Profiling and Bioassay-Guided Fractionation of Zataria multiflora Boiss. Hydroethanolic Leaf Extracts for Identification of Broad-Spectrum Pre and Postharvest Antifungal Agents. Molecules. 2022; 27(24):8903. https://doi.org/10.3390/molecules27248903
Chicago/Turabian StyleKarimi, Ali, Torsten Meiners, and Christoph Böttcher. 2022. "Metabolite Profiling and Bioassay-Guided Fractionation of Zataria multiflora Boiss. Hydroethanolic Leaf Extracts for Identification of Broad-Spectrum Pre and Postharvest Antifungal Agents" Molecules 27, no. 24: 8903. https://doi.org/10.3390/molecules27248903