
Proteomic Insights into Cardiac Fibrosis: From Pathophysiological Mechanisms to Therapeutic Opportunities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fundamentals of Cardiac Fibrosis
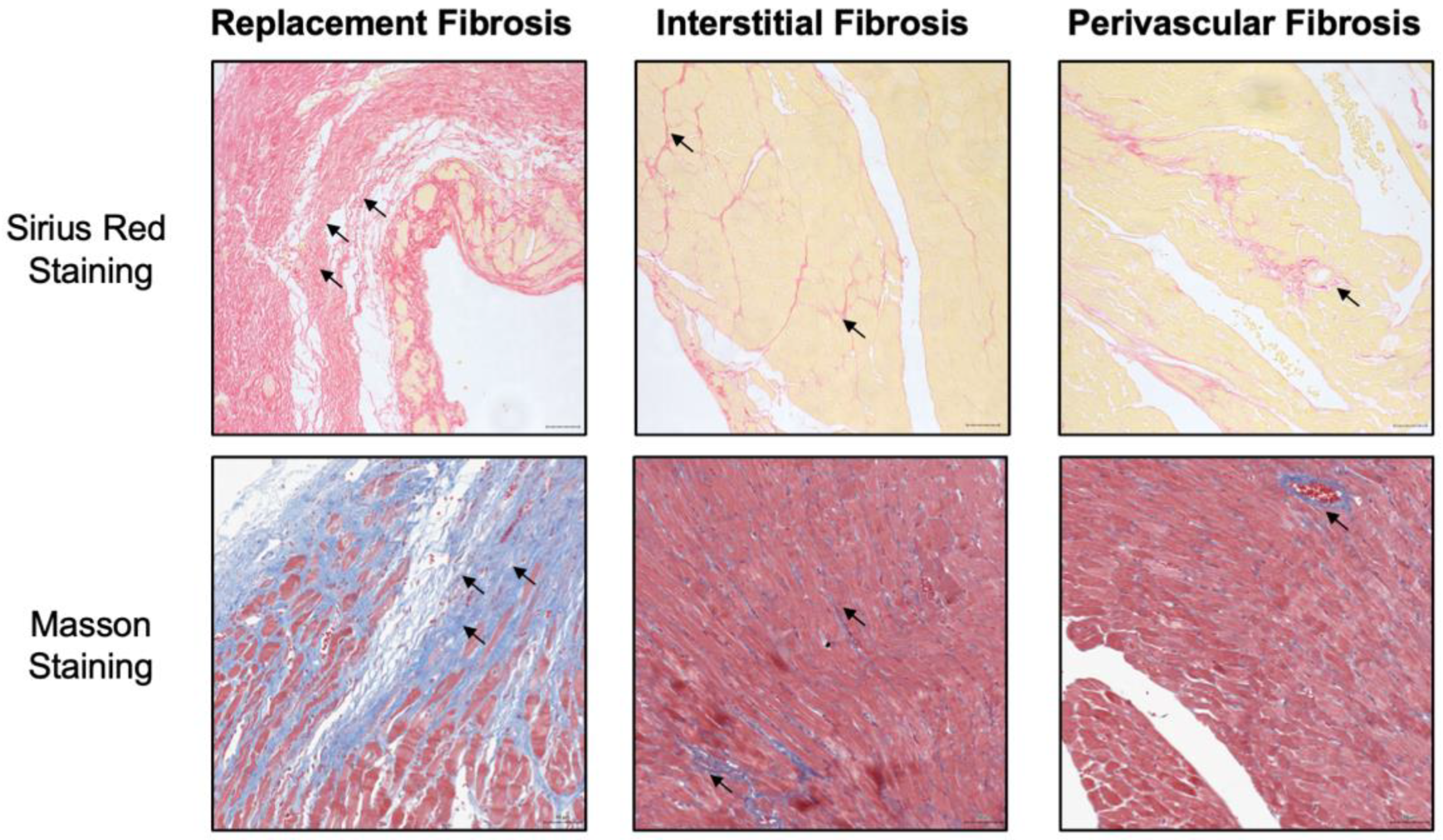
2.1. The Histopathological Types of Cardiac Fibrosis
2.2. Imaging and Biochemical Methods to Detect Cardiac Fibrosis
2.3. Emerging Therapeutic Strategies for Cardiac Fibrosis
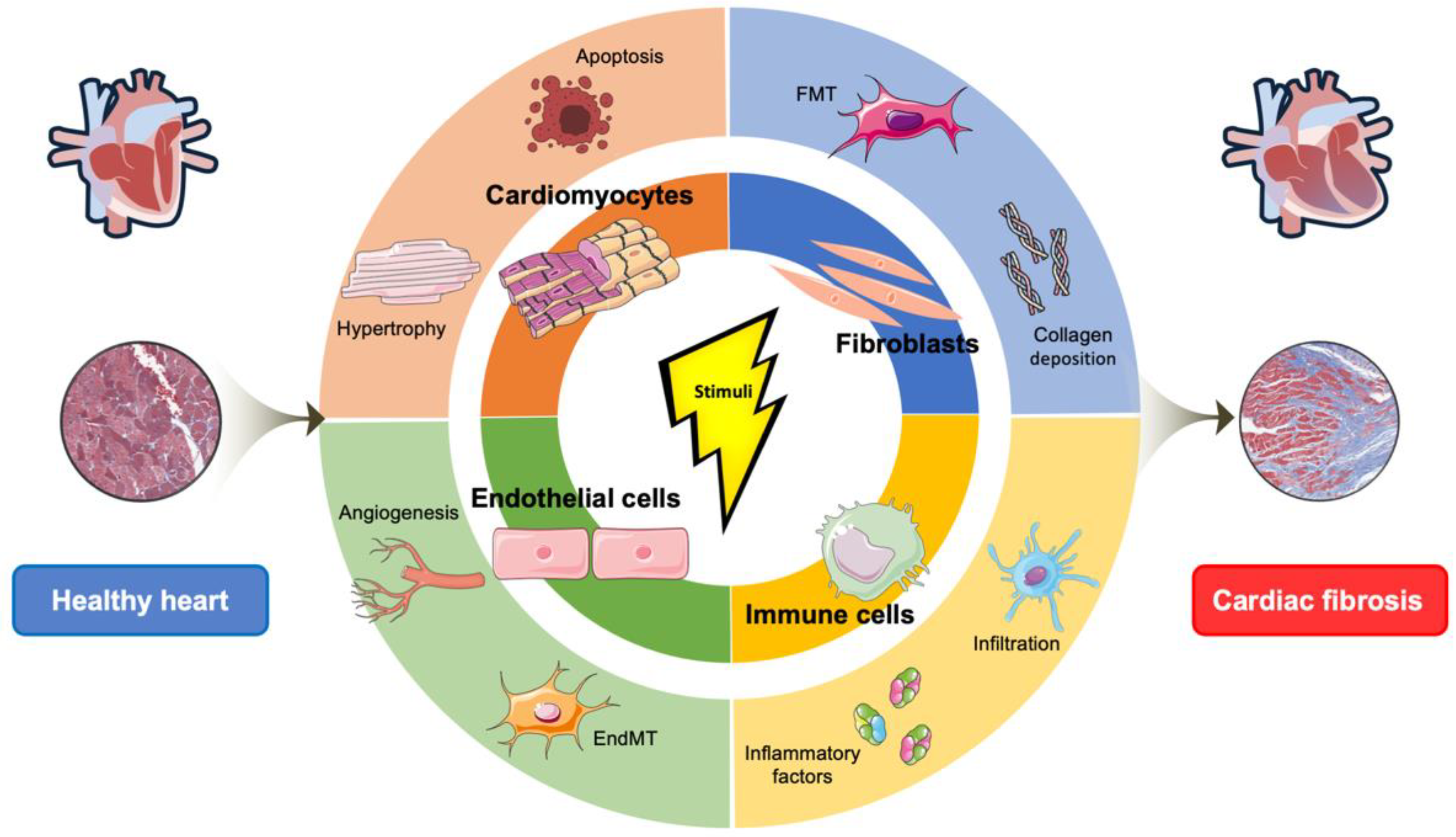
3. Cell Biological Mechanisms Underlining Cardiac Fibrosis
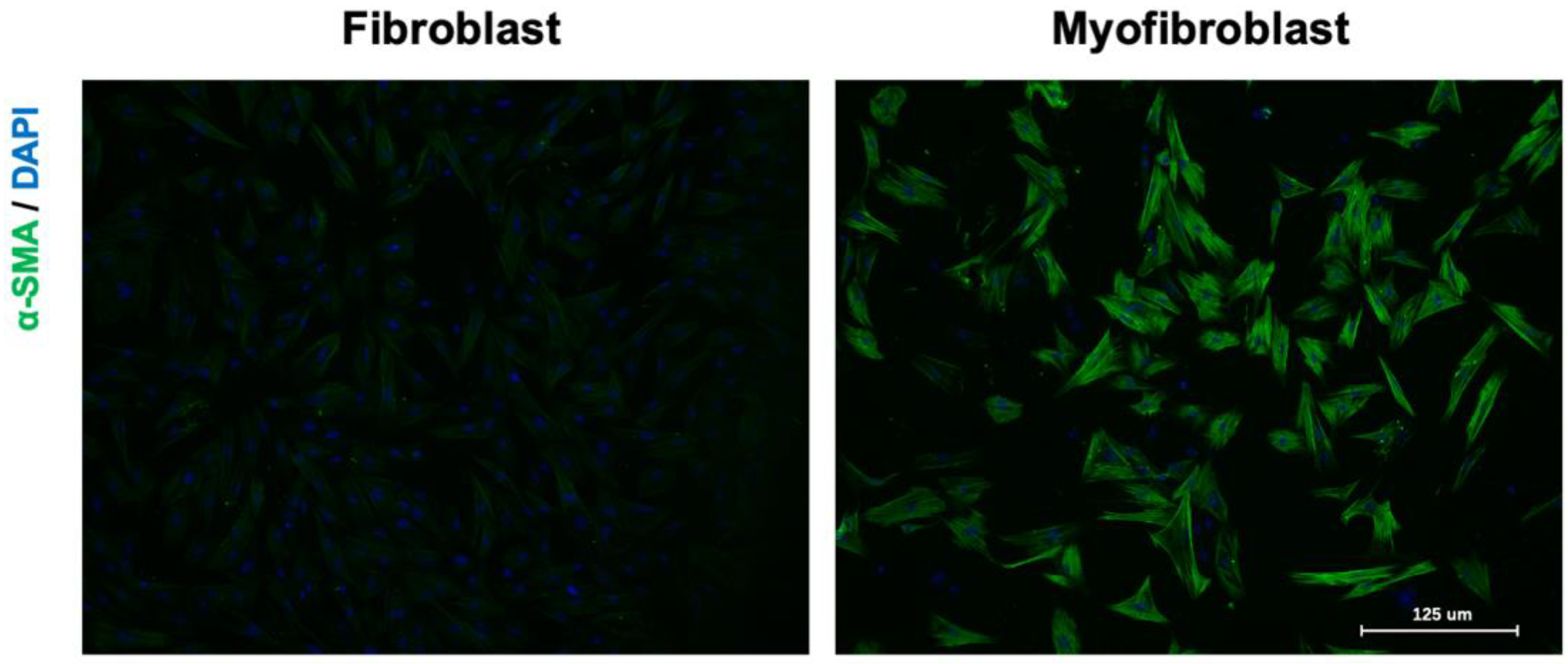
3.1. The Cellular Effectors of Cardiac Fibrosis
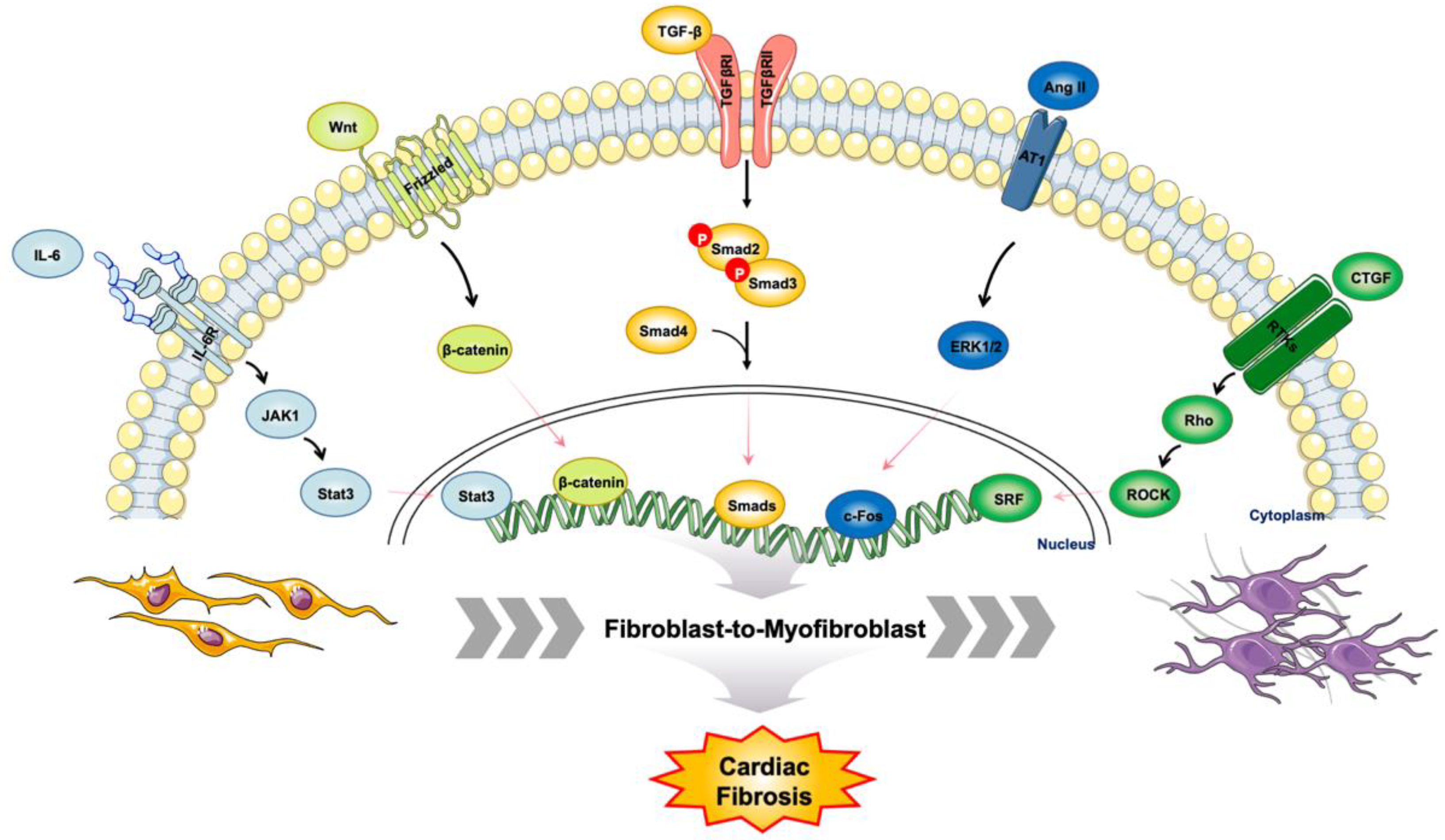
3.2. The Master Regulators of Cardiac Fibrosis
3.3. Progress on Proteomic Studies of Patients with Cardiovascular Disease
4. Proteomic Insights into Cardiac Fibrosis
4.1. Understanding Cardiac Fibrosis in Animal Models
4.2. Region and Cell-Type Resolved Quantitative Proteomic Analysis of Cardiac Fibrosis
4.2.1. Cardiomyocytes
4.2.2. Cardiac Fibroblasts
4.2.3. Endothelial Cells
4.2.4. Extracellular Matrix Remodeling
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 2019, 65, 70–99. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; González, A.; Pizard, A.; Papageorgiou, A.P.; López-Andrés, N.; Jaisser, F.; Thum, T.; Zannad, F.; Díez, J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur. J. Heart Fail. 2015, 17, 764–771. [Google Scholar] [CrossRef] [Green Version]
- González, A.; Schelbert, E.B.; Díez, J.; Butler, J. Myocardial Interstitial Fibrosis in Heart Failure: Biological and Translational Perspectives. J. Am. Coll. Cardiol. 2018, 71, 1696–1706. [Google Scholar] [CrossRef]
- Weber, K.T.; Janicki, J.S.; Shroff, S.G.; Pick, R.; Chen, R.M.; Bashey, R.I. Collagen remodeling of the pressure-overloaded, hypertrophied nonhuman primate myocardium. Circ. Res. 1988, 62, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Frangogiannis, N.G. Cardiac fibrosis. Cardiovasc. Res. 2021, 117, 1450–1488. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Francis Stuart, S.D.; De Jesus, N.M.; Lindsey, M.L.; Ripplinger, C.M. The crossroads of inflammation, fibrosis, and arrhythmia following myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 114–122. [Google Scholar] [CrossRef]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borer, J.S.; Truter, S.; Herrold, E.M.; Falcone, D.J.; Pena, M.; Carter, J.N.; Dumlao, T.F.; Lee, J.A.; Supino, P.G. Myocardial fibrosis in chronic aortic regurgitation: Molecular and cellular responses to volume overload. Circulation 2002, 105, 1837–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar]
- Van Heerebeek, L.; Hamdani, N.; Handoko, M.L.; Falcao-Pires, I.; Musters, R.J.; Kupreishvili, K.; Ijsselmuiden, A.J.; Schalkwijk, C.G.; Bronzwaer, J.G.; Diamant, M.; et al. Diastolic stiffness of the failing diabetic heart: Importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation 2008, 117, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalera, M.; Wang, J.; Frangogiannis, N.G. Obesity, metabolic dysfunction, and cardiac fibrosis: Pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl. Res. 2014, 164, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Bovelli, D.; Plataniotis, G.; Roila, F. Cardiotoxicity of chemotherapeutic agents and radiotherapy-related heart disease: ESMO Clinical Practice Guidelines. Ann. Oncol. 2010, 21 (Suppl. 5), v277–v282. [Google Scholar] [CrossRef]
- Richards, D.A.; Aronovitz, M.J.; Calamaras, T.D.; Tam, K.; Martin, G.L.; Liu, P.; Bowditch, H.K.; Zhang, P.; Huggins, G.S.; Blanton, R.M. Distinct Phenotypes Induced by Three Degrees of Transverse Aortic Constriction in Mice. Sci. Rep. 2019, 9, 5844. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.P.; Qu, Z.; Weiss, J.N. Cardiac fibrosis and arrhythmogenesis: The road to repair is paved with perils. J. Mol. Cell. Cardiol. 2014, 70, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Cooper, L.T.; Baughman, K.L.; Feldman, A.M.; Frustaci, A.; Jessup, M.; Kuhl, U.; Levine, G.N.; Narula, J.; Starling, R.C.; Towbin, J.; et al. The role of endomyocardial biopsy in the management of cardiovascular disease: A scientific statement from the American Heart Association, the American College of Cardiology, and the European Society of Cardiology. Endorsed by the Heart Failure Society of America and the Heart Failure Association of the European Society of Cardiology. J. Am. Coll. Cardiol. 2007, 50, 1914–1931. [Google Scholar]
- Martos, R.; Baugh, J.; Ledwidge, M.; O’Loughlin, C.; Conlon, C.; Patle, A.; Donnelly, S.C.; McDonald, K. Diastolic heart failure: Evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation 2007, 115, 888–895. [Google Scholar] [CrossRef] [Green Version]
- Hahn, V.S.; Yanek, L.R.; Vaishnav, J.; Ying, W.; Vaidya, D.; Lee, Y.Z.J.; Riley, S.J.; Subramanya, V.; Brown, E.E.; Hopkins, C.D.; et al. Endomyocardial Biopsy Characterization of Heart Failure With Preserved Ejection Fraction and Prevalence of Cardiac Amyloidosis. JACC Heart Fail. 2020, 8, 712–724. [Google Scholar] [CrossRef] [PubMed]
- Gyöngyösi, M.; Winkler, J.; Ramos, I.; Do, Q.T.; Firat, H.; McDonald, K.; González, A.; Thum, T.; Díez, J.; Jaisser, F.; et al. Myocardial fibrosis: Biomedical research from bench to bedside. Eur. J. Heart Fail. 2017, 19, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Jellis, C.; Martin, J.; Narula, J.; Marwick, T.H. Assessment of nonischemic myocardial fibrosis. J. Am. Coll. Cardiol. 2010, 56, 89–97. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Ge, Y.; Singh, A.; Grani, C.; Kwong, R.Y. Multimodality Imaging Assessment of Myocardial Fibrosis. JACC Cardiovasc. Imaging 2021, 14, 2457–2469. [Google Scholar] [CrossRef]
- Graham-Brown, M.P.; Patel, A.S.; Stensel, D.J.; March, D.S.; Marsh, A.M.; McAdam, J.; McCann, G.P.; Burton, J.O. Imaging of Myocardial Fibrosis in Patients with End-Stage Renal Disease: Current Limitations and Future Possibilities. Biomed Res. Int. 2017, 2017, 5453606. [Google Scholar] [CrossRef] [Green Version]
- Mewton, N.; Liu, C.Y.; Croisille, P.; Bluemke, D.; Lima, J.A. Assessment of myocardial fibrosis with cardiovascular magnetic resonance. J. Am. Coll. Cardiol. 2011, 57, 891–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambale-Venkatesh, B.; Lima, J.A. Cardiac MRI: A central prognostic tool in myocardial fibrosis. Nat. Rev. Cardiol. 2015, 12, 18–29. [Google Scholar] [CrossRef]
- López, B.; González, A.; Ravassa, S.; Beaumont, J.; Moreno, M.U.; San José, G.; Querejeta, R.; Díez, J. Circulating Biomarkers of Myocardial Fibrosis: The Need for a Reappraisal. J. Am. Coll. Cardiol. 2015, 65, 2449–2456. [Google Scholar] [CrossRef] [Green Version]
- Querejeta, R.; López, B.; González, A.; Sánchez, E.; Larman, M.; Martínez Ubago, J.L.; Díez, J. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: Relation to myocardial fibrosis. Circulation 2004, 110, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Raafs, A.G.; Verdonschot, J.A.J.; Henkens, M.; Adriaans, B.P.; Wang, P.; Derks, K.; Abdul Hamid, M.A.; Knackstedt, C.; van Empel, V.P.M.; Díez, J.; et al. The combination of carboxy-terminal propeptide of procollagen type I blood levels and late gadolinium enhancement at cardiac magnetic resonance provides additional prognostic information in idiopathic dilated cardiomyopathy—A multilevel assessment of myocardial fibrosis in dilated cardiomyopathy. Eur. J. Heart Fail. 2021, 23, 933–944. [Google Scholar]
- Park, S.; Ranjbarvaziri, S.; Zhao, P.; Ardehali, R. Cardiac Fibrosis Is Associated With Decreased Circulating Levels of Full-Length CILP in Heart Failure. JACC Basic. Transl. Sci. 2020, 5, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Vértes, V.; Porpáczy, A.; Nógrádi, Á.; Tőkés-Füzesi, M.; Hajdu, M.; Czirják, L.; Komócsi, A.; Faludi, R. Galectin-3 and sST2: Associations to the echocardiographic markers of the myocardial mechanics in systemic sclerosis—A pilot study. Cardiovasc. Ultrasound 2022, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Shi, N.; Wang, Z.; Zhu, H.; Liu, W.; Zhao, M.; Jiang, X.; Zhao, J.; Ren, C.; Zhang, Y.; Luo, L. Research progress on drugs targeting the TGF-beta signaling pathway in fibrotic diseases. Immunol. Res. 2022, 70, 276–288. [Google Scholar] [CrossRef] [PubMed]
- AlQudah, M.; Hale, T.M.; Czubryt, M.P. Targeting the renin-angiotensin-aldosterone system in fibrosis. Matrix Biol. 2020, 91–92, 92–108. [Google Scholar] [CrossRef]
- Akhurst, R.J.; Hata, A. Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [Green Version]
- Parichatikanond, W.; Luangmonkong, T.; Mangmool, S.; Kurose, H. Therapeutic Targets for the Treatment of Cardiac Fibrosis and Cancer: Focusing on TGF-β Signaling. Front. Cardiovasc. Med. 2020, 7, 34. [Google Scholar] [CrossRef]
- Yao, Y.; Li, A.; Wang, S.; Lu, Y.; Xie, J.; Zhang, H.; Zhang, D.; Ding, J.; Wang, Z.; Tu, C.; et al. Multifunctional elastomer cardiac patches for preventing left ventricle remodeling after myocardial infarction in vivo. Biomaterials 2022, 282, 121382. [Google Scholar] [CrossRef]
- Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Worth, C.L.; Lindberg, E.L.; Kanda, M.; Polanski, K.; Heinig, M.; Lee, M.; et al. Cells of the adult human heart. Nature 2020, 588, 466–472. [Google Scholar] [CrossRef]
- Tucker, N.R.; Chaffin, M.; Fleming, S.J.; Hall, A.W.; Parsons, V.A.; Bedi, K.C., Jr.; Akkad, A.D.; Herndon, C.N.; Arduini, A.; Papangeli, I.; et al. Transcriptional and Cellular Diversity of the Human Heart. Circulation 2020, 142, 466–482. [Google Scholar] [CrossRef]
- Wang, L.; Yu, P.; Zhou, B.; Song, J.; Li, Z.; Zhang, M.; Guo, G.; Wang, Y.; Chen, X.; Han, L.; et al. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat. Cell. Biol. 2020, 22, 108–119. [Google Scholar] [CrossRef]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Lazaropoulos, M.P.; Elrod, J.W. Myofibroblasts and Fibrosis: Mitochondrial and Metabolic Control of Cellular Differentiation. Circ. Res. 2020, 127, 427–447. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.; Gehmlich, K.; Denning, C.; Pavlovic, D. Complex Relationship Between Cardiac Fibroblasts and Cardiomyocytes in Health and Disease. J. Am. Heart Assoc. 2021, 10, e019338. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/βcatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. Embo J. 2012, 31, 429–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Linthout, S.; Miteva, K.; Tschope, C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc. Res. 2014, 102, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Carretero, O.A.; Liao, T.D.; Peng, H.; Shesely, E.G.; Xu, J.; Liu, T.S.; Yang, J.J.; Reudelhuber, T.L.; Yang, X.P. Local angiotensin II aggravates cardiac remodeling in hypertension. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1328–H1338. [Google Scholar] [CrossRef] [Green Version]
- Kawano, H.; Do, Y.S.; Kawano, Y.; Starnes, V.; Barr, M.; Law, R.E.; Hsueh, W.A. Angiotensin II has multiple profibrotic effects in human cardiac fibroblasts. Circulation 2000, 101, 1130–1137. [Google Scholar] [CrossRef] [Green Version]
- Lyu, L.; Wang, H.; Li, B.; Qin, Q.; Qi, L.; Nagarkatti, M.; Nagarkatti, P.; Janicki, J.S.; Wang, X.L.; Cui, T. A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes. J. Mol. Cell. Cardiol. 2015, 89, 268–279. [Google Scholar] [CrossRef] [Green Version]
- Campbell, S.E.; Katwa, L.C. Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J. Mol. Cell. Cardiol. 1997, 29, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem. Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-β1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed]
- Groppe, J.; Hinck, C.S.; Samavarchi-Tehrani, P.; Zubieta, C.; Schuermann, J.P.; Taylor, A.B.; Schwarz, P.M.; Wrana, J.L.; Hinck, A.P. Cooperative assembly of TGF-beta superfamily signaling complexes is mediated by two disparate mechanisms and distinct modes of receptor binding. Mol. Cell. 2008, 29, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β Mediated SMAD Signaling for the Prevention of Fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Yang, T.; Lu, D.W.; Zhao, H.; Feng, Y.L.; Chen, H.; Chen, D.Q.; Vaziri, N.D.; Zhao, Y.Y. Central role of dysregulation of TGF-β/Smad in CKD progression and potential targets of its treatment. Biomed Pharm. 2018, 101, 670–681. [Google Scholar] [CrossRef] [Green Version]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Transforming growth factor-β in myocardial disease. Nat. Rev. Cardiol. 2022, 19, 435–455. [Google Scholar] [CrossRef]
- Pandey, A.; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837–846. [Google Scholar] [CrossRef]
- Larance, M.; Lamond, A.I. Multidimensional proteomics for cell biology. Nat. Rev. Mol. Cell Biol. 2015, 16, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef]
- Schoof, E.M.; Furtwangler, B.; Uresin, N.; Rapin, N.; Savickas, S.; Gentil, C.; Lechman, E.; Keller, U.A.D.; Dick, J.E.; Porse, B.T. Quantitative single-cell proteomics as a tool to characterize cellular hierarchies. Nat. Commun. 2021, 12, 3341. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, D.; Jarnuczak, A.F.; Vieitez, C.; Gehre, M.; Soucheray, M.; Mateus, A.; Kleefeldt, A.A.; Hill, A.; Garcia-Alonso, L.; Stein, F.; et al. The functional landscape of the human phosphoproteome. Nat. Biotechnol. 2020, 38, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Hammaren, H.M.; Geissen, E.M.; Potel, C.M.; Beck, M.; Savitski, M.M. Protein-Peptide Turnover Profiling reveals the order of PTM addition and removal during protein maturation. Nat. Commun. 2022, 13, 7431. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 2021, 184, 3022–3040.e28. [Google Scholar] [CrossRef] [PubMed]
- Keshishian, H.; Burgess, M.W.; Specht, H.; Wallace, L.; Clauser, K.R.; Gillette, M.A.; Carr, S.A. Quantitative, multiplexed workflow for deep analysis of human blood plasma and biomarker discovery by mass spectrometry. Nat. Protoc. 2017, 12, 1683–1701. [Google Scholar] [CrossRef]
- Mertins, P.; Tang, L.C.; Krug, K.; Clark, D.J.; Gritsenko, M.A.; Chen, L.; Clauser, K.R.; Clauss, T.R.; Shah, P.; Gillette, M.A.; et al. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13, 1632–1661. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Frank, M.; Ha, A.; Bludau, I.; Voytik, E.; Kaspar-Schoenefeld, S.; Lubeck, M.; Raether, O.; Bache, N.; et al. diaPASEF: Parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat. Methods 2020, 17, 1229–1236. [Google Scholar] [CrossRef]
- Meier, F.; Brunner, A.D.; Koch, S.; Koch, H.; Lubeck, M.; Krause, M.; Goedecke, N.; Decker, J.; Kosinski, T.; Park, M.A.; et al. Online Parallel Accumulation-Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer. Mol. Cell. Proteom. 2018, 17, 2534–2545. [Google Scholar] [CrossRef] [Green Version]
- De Boer, R.A.; De Keulenaer, G.; Bauersachs, J.; Brutsaert, D.; Cleland, J.G.; Diez, J.; Du, X.J.; Ford, P.; Heinzel, F.R.; Lipson, K.E.; et al. Towards better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 272–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, M.Y.; Efthymios, M.; Tan, S.H.; Pickering, J.W.; Troughton, R.; Pemberton, C.; Ho, H.H.; Prabath, J.F.; Drum, C.L.; Ling, L.H.; et al. Prioritizing Candidates of Post-Myocardial Infarction Heart Failure Using Plasma Proteomics and Single-Cell Transcriptomics. Circulation 2020, 142, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Ortega, F.B.; Lavie, C.J.; Blair, S.N. Obesity and Cardiovascular Disease. Circ. Res. 2016, 118, 1752–1770. [Google Scholar] [CrossRef] [PubMed]
- Kresoja, K.P.; Rommel, K.P.; Wachter, R.; Henger, S.; Besler, C.; Klöting, N.; Schnelle, M.; Hoffmann, A.; Büttner, P.; Ceglarek, U.; et al. Proteomics to improve phenotyping in obese patients with heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2021, 23, 1633–1644. [Google Scholar] [CrossRef]
- Swinnen, M.; Vanhoutte, D.; Van Almen, G.C.; Hamdani, N.; Schellings, M.W.; D’Hooge, J.; Van der Velden, J.; Weaver, M.S.; Sage, E.H.; Bornstein, P.; et al. Absence of thrombospondin-2 causes age-related dilated cardiomyopathy. Circulation 2009, 120, 1585–1597. [Google Scholar] [CrossRef] [Green Version]
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovasc. Res. 2019, 115, 1838–1849. [Google Scholar] [CrossRef]
- Gao, E.; Lei, Y.H.; Shang, X.; Huang, Z.M.; Zuo, L.; Boucher, M.; Fan, Q.; Chuprun, J.K.; Ma, X.L.; Koch, W.J. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 2010, 107, 1445–1453. [Google Scholar] [CrossRef]
- Li, Y.; Wang, C.; Li, T.; Ma, L.; Fan, F.; Jin, Y.; Shen, J. The whole transcriptome and proteome changes in the early stage of myocardial infarction. Cell Death Discov. 2019, 5, 73. [Google Scholar] [CrossRef] [Green Version]
- Gueugneau, M.; d’Hose, D.; Barbe, C.; de Barsy, M.; Lause, P.; Maiter, D.; Bindels, L.B.; Delzenne, N.M.; Schaeffer, L.; Gangloff, Y.G.; et al. Increased Serpina3n release into circulation during glucocorticoid-mediated muscle atrophy. J. Cachexia Sarcopenia Muscle 2018, 9, 929–946. [Google Scholar] [CrossRef] [Green Version]
- Cameron, V.A.; Aitken, G.D.; Ellmers, L.J.; Kennedy, M.A.; Espiner, E.A. The sites of gene expression of atrial, brain, and C-type natriuretic peptides in mouse fetal development: Temporal changes in embryos and placenta. Endocrinology 1996, 137, 817–824. [Google Scholar] [CrossRef]
- Horsthuis, T.; Houweling, A.C.; Habets, P.E.; de Lange, F.J.; el Azzouzi, H.; Clout, D.E.; Moorman, A.F.; Christoffels, V.M. Distinct regulation of developmental and heart disease-induced atrial natriuretic factor expression by two separate distal sequences. Circ. Res. 2008, 102, 849–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, J.C.K.; van Duijvenboden, K.; Krijger, P.H.L.; Hooijkaas, I.B.; van der Made, I.; de Gier-de Vries, C.; Wakker, V.; Creemers, E.E.; de Laat, W.; Boukens, B.J.; et al. Genetic Dissection of a Super Enhancer Controlling the Nppa-Nppb Cluster in the Heart. Circ. Res. 2021, 128, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gao, W.; Li, L.; Hao, J.; Yang, B.; Wang, T.; Li, L.; Bai, X.; Li, F.; Ren, H.; et al. Annexin A1 protects against cerebral ischemia-reperfusion injury by modulating microglia/macrophage polarization via FPR2/ALX-dependent AMPK-mTOR pathway. J. Neuroinflamm. 2021, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Rockman, H.A.; Ross, R.S.; Harris, A.N.; Knowlton, K.U.; Steinhelper, M.E.; Field, L.J.; Ross, J., Jr.; Chien, K.R. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 1991, 88, 8277–8281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida, A.C.; van Oort, R.J.; Wehrens, X.H. Transverse aortic constriction in mice. J. Vis. Exp. 2010, 38, e1729. [Google Scholar]
- Li, X.; Zhang, W.; Cao, Q.; Wang, Z.; Zhao, M.; Xu, L.; Zhuang, Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef] [Green Version]
- Norton, G.R.; Woodiwiss, A.J.; Gaasch, W.H.; Mela, T.; Chung, E.S.; Aurigemma, G.P.; Meyer, T.E. Heart failure in pressure overload hypertrophy. The relative roles of ventricular remodeling and myocardial dysfunction. J. Am. Coll. Cardiol. 2002, 39, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Van den Bosch, B.J.; Lindsey, P.J.; van den Burg, C.M.; van der Vlies, S.A.; Lips, D.J.; van der Vusse, G.J.; Ayoubi, T.A.; Doevendans, P.A.; Smeets, H.J. Early and transient gene expression changes in pressure overload-induced cardiac hypertrophy in mice. Genomics 2006, 88, 480–488. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lian, H.; Yu, J.; Wu, J.; Chen, X.; Wang, P.; Tian, L.; Yang, Y.; Yang, J.; Li, D.; et al. Study on diverse pathological characteristics of heart failure in different stages based on proteomics. J. Cell. Mol. Med. 2022, 26, 1169–1182. [Google Scholar] [CrossRef]
- Hasselberg, N.E.; Haland, T.F.; Saberniak, J.; Brekke, P.H.; Berge, K.E.; Leren, T.P.; Edvardsen, T.; Haugaa, K.H. Lamin A/C cardiomyopathy: Young onset, high penetrance, and frequent need for heart transplantation. Eur. Heart J. 2018, 39, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogenhof, M.M.G.; Beqqali, A.; Amin, A.S.; van der Made, I.; Aufiero, S.; Khan, M.A.F.; Schumacher, C.A.; Jansweijer, J.A.; van Spaendonck-Zwarts, K.Y.; Remme, C.A.; et al. RBM20 Mutations Induce an Arrhythmogenic Dilated Cardiomyopathy Related to Disturbed Calcium Handling. Circulation 2018, 138, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Coats, C.J.; Heywood, W.E.; Virasami, A.; Ashrafi, N.; Syrris, P.; Dos Remedios, C.; Treibel, T.A.; Moon, J.C.; Lopes, L.R.; McGregor, C.G.A.; et al. Proteomic Analysis of the Myocardium in Hypertrophic Obstructive Cardiomyopathy. Circ. Genom. Precis. Med. 2018, 11, e001974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Parker, B.L.; Pearson, E.; Hunter, B.; Cao, J.; Koay, Y.C.; Guneratne, O.; James, D.E.; Yang, J.; Lal, S.; et al. Core functional nodes and sex-specific pathways in human ischaemic and dilated cardiomyopathy. Nat. Commun. 2020, 11, 2843. [Google Scholar] [CrossRef] [PubMed]
- Herrington, D.M.; Mao, C.; Parker, S.J.; Fu, Z.; Yu, G.; Chen, L.; Venkatraman, V.; Fu, Y.; Wang, Y.; Howard, T.D.; et al. Proteomic Architecture of Human Coronary and Aortic Atherosclerosis. Circulation 2018, 137, 2741–2756. [Google Scholar] [CrossRef]
- Doll, S.; Dreßen, M.; Geyer, P.E.; Itzhak, D.N.; Braun, C.; Doppler, S.A.; Meier, F.; Deutsch, M.A.; Lahm, H.; Lange, R.; et al. Region and cell-type resolved quantitative proteomic map of the human heart. Nat. Commun. 2017, 8, 1469. [Google Scholar] [CrossRef] [Green Version]
- Linscheid, N.; Poulsen, P.C.; Pedersen, I.D.; Gregers, E.; Svendsen, J.H.; Olesen, M.S.; Olsen, J.V.; Delmar, M.; Lundby, A. Quantitative Proteomics of Human Heart Samples Collected In Vivo Reveal the Remodeled Protein Landscape of Dilated Left Atrium without Atrial Fibrillation. Mol. Cell. Proteom. 2020, 19, 1132–1144. [Google Scholar] [CrossRef] [Green Version]
- Nippert, F.; Schreckenberg, R.; Schlüter, K.D. Isolation and Cultivation of Adult Rat Cardiomyocytes. J. Vis. Exp. 2017, 128, e56634. [Google Scholar] [CrossRef]
- Wu, K.; Li, L.L.; Li, Y.K.; Peng, X.D.; Zhang, M.X.; Liu, K.S.; Wang, X.S.; Yang, J.X.; Wen, S.N.; Ruan, Y.F.; et al. Modifications of the Langendorff Method for Simultaneous Isolation of Atrial and Ventricular Myocytes from Adult Mice. J. Vis. Exp. 2021, 171, e62514. [Google Scholar] [CrossRef]
- Gündüz, D.; Hamm, C.W.; Aslam, M. Simultaneous Isolation of High Quality Cardiomyocytes, Endothelial Cells, and Fibroblasts from an Adult Rat Heart. J. Vis. Exp. 2017, 123, e55601. [Google Scholar] [CrossRef]
- Melzer, M.; Beier, D.; Young, P.P.; Saraswati, S. Isolation and Characterization of Adult Cardiac Fibroblasts and Myofibroblasts. J. Vis. Exp. 2020, 157, e60909. [Google Scholar] [CrossRef] [PubMed]
- Wojtkiewicz, M.; Berg Luecke, L.; Castro, C.; Burkovetskaya, M.; Mesidor, R.; Gundry, R.L. Bottom-up proteomic analysis of human adult cardiac tissue and isolated cardiomyocytes. J. Mol. Cell. Cardiol. 2022, 162, 20–31. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Ma, Q.; Xie, W.; Liou, C.L.; Zhang, D.; Sweat, M.E.; Jardin, B.D.; Naya, F.J.; Guo, Y.; Cheng, H.; et al. CMYA5 establishes cardiac dyad architecture and positioning. Nat. Commun. 2022, 13, 2185. [Google Scholar] [CrossRef] [PubMed]
- Tirziu, D.; Giordano, F.J.; Simons, M. Cell communications in the heart. Circulation 2010, 122, 928–937. [Google Scholar] [CrossRef]
- Wagh, K.; Ishikawa, M.; Garcia, D.A.; Stavreva, D.A.; Upadhyaya, A.; Hager, G.L. Mechanical Regulation of Transcription: Recent Advances. Trends Cell Biol. 2021, 31, 457–472. [Google Scholar] [CrossRef]
- Kuhn, T.C.; Knobel, J.; Burkert-Rettenmaier, S.; Li, X.; Meyer, I.S.; Jungmann, A.; Sicklinger, F.; Backs, J.; Lasitschka, F.; Muller, O.J.; et al. Secretome Analysis of Cardiomyocytes Identifies PCSK6 (Proprotein Convertase Subtilisin/Kexin Type 6) as a Novel Player in Cardiac Remodeling After Myocardial Infarction. Circulation 2020, 141, 1628–1644. [Google Scholar] [CrossRef]
- Shah, H.; Hacker, A.; Langburt, D.; Dewar, M.; McFadden, M.J.; Zhang, H.; Kuzmanov, U.; Zhou, Y.Q.; Hussain, B.; Ehsan, F.; et al. Myocardial Infarction Induces Cardiac Fibroblast Transformation within Injured and Noninjured Regions of the Mouse Heart. J. Proteome Res. 2021, 20, 2867–2881. [Google Scholar] [CrossRef]
- Zhang, Q.J.; He, Y.; Li, Y.; Shen, H.; Lin, L.; Zhu, M.; Wang, Z.; Luo, X.; Hill, J.A.; Cao, D.; et al. Matricellular Protein Cilp1 Promotes Myocardial Fibrosis in Response to Myocardial Infarction. Circ. Res. 2021, 129, 1021–1035. [Google Scholar] [CrossRef]
- Kalyanasundaram, A.; Li, N.; Gardner, M.L.; Artiga, E.J.; Hansen, B.J.; Webb, A.; Freitas, M.A.; Pietrzak, M.; Whitson, B.A.; Mokadam, N.A.; et al. Fibroblast-Specific Proteotranscriptomes Reveal Distinct Fibrotic Signatures of Human Sinoatrial Node in Nonfailing and Failing Hearts. Circulation 2021, 144, 126–143. [Google Scholar] [CrossRef]
- Dittrich, G.M.; Froese, N.; Wang, X.; Kroeger, H.; Wang, H.; Szaroszyk, M.; Malek-Mohammadi, M.; Cordero, J.; Keles, M.; Korf-Klingebiel, M.; et al. Fibroblast GATA-4 and GATA-6 promote myocardial adaptation to pressure overload by enhancing cardiac angiogenesis. Basic Res. Cardiol. 2021, 116, 26. [Google Scholar] [CrossRef]
- Sun, X.; Nkennor, B.; Mastikhina, O.; Soon, K.; Nunes, S.S. Endothelium-mediated contributions to fibrosis. Semin. Cell. Dev. Biol. 2020, 101, 78–86. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell. Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvandi, Z.; Bischoff, J. Endothelial-Mesenchymal Transition in Cardiovascular Disease. Arter. Thromb. Vasc. Biol. 2021, 41, 2357–2369. [Google Scholar] [CrossRef] [PubMed]
- Trenson, S.; Hermans, H.; Craps, S.; Pokreisz, P.; de Zeeuw, P.; Van Wauwe, J.; Gillijns, H.; Veltman, D.; Wei, F.; Caluwe, E.; et al. Cardiac Microvascular Endothelial Cells in Pressure Overload-Induced Heart Disease. Circ. Heart Fail. 2021, 14, e006979. [Google Scholar] [CrossRef]
- Garcia, V.; Park, E.J.; Siragusa, M.; Frohlich, F.; Mahfuzul Haque, M.; Pascale, J.V.; Heberlein, K.R.; Isakson, B.E.; Stuehr, D.J.; Sessa, W.C. Unbiased proteomics identifies plasminogen activator inhibitor-1 as a negative regulator of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 2020, 117, 9497–9507. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. The extracellular matrix in myocardial injury, repair, and remodeling. J. Clin. Investig. 2017, 127, 1600–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, S.H.; Mouton, A.J.; DeLeon-Pennell, K.Y.; Genovese, F.; Karsdal, M.; Lindsey, M.L. Understanding cardiac extracellular matrix remodeling to develop biomarkers of myocardial infarction outcomes. Matrix Biol. 2019, 75–76, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Barallobre-Barreiro, J.; Radovits, T.; Fava, M.; Mayr, U.; Lin, W.Y.; Ermolaeva, E.; Martinez-Lopez, D.; Lindberg, E.L.; Duregotti, E.; Daroczi, L.; et al. Extracellular Matrix in Heart Failure: Role of ADAMTS5 in Proteoglycan Remodeling. Circulation 2021, 144, 2021–2034. [Google Scholar] [CrossRef] [PubMed]
- Barallobre-Barreiro, J.; Didangelos, A.; Schoendube, F.A.; Drozdov, I.; Yin, X.; Fernandez-Caggiano, M.; Willeit, P.; Puntmann, V.O.; Aldama-Lopez, G.; Shah, A.M.; et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation 2012, 125, 789–802. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, R.; Lin, E.; Song, J.; Wang, Y.; Lin, L. Proteomic Insights into Cardiac Fibrosis: From Pathophysiological Mechanisms to Therapeutic Opportunities. Molecules 2022, 27, 8784. https://doi.org/10.3390/molecules27248784
Qi R, Lin E, Song J, Wang Y, Lin L. Proteomic Insights into Cardiac Fibrosis: From Pathophysiological Mechanisms to Therapeutic Opportunities. Molecules. 2022; 27(24):8784. https://doi.org/10.3390/molecules27248784
Chicago/Turabian StyleQi, Ruiqiang, E. Lin, Juan Song, Yan Wang, and Ling Lin. 2022. "Proteomic Insights into Cardiac Fibrosis: From Pathophysiological Mechanisms to Therapeutic Opportunities" Molecules 27, no. 24: 8784. https://doi.org/10.3390/molecules27248784