3.5. Synthesis
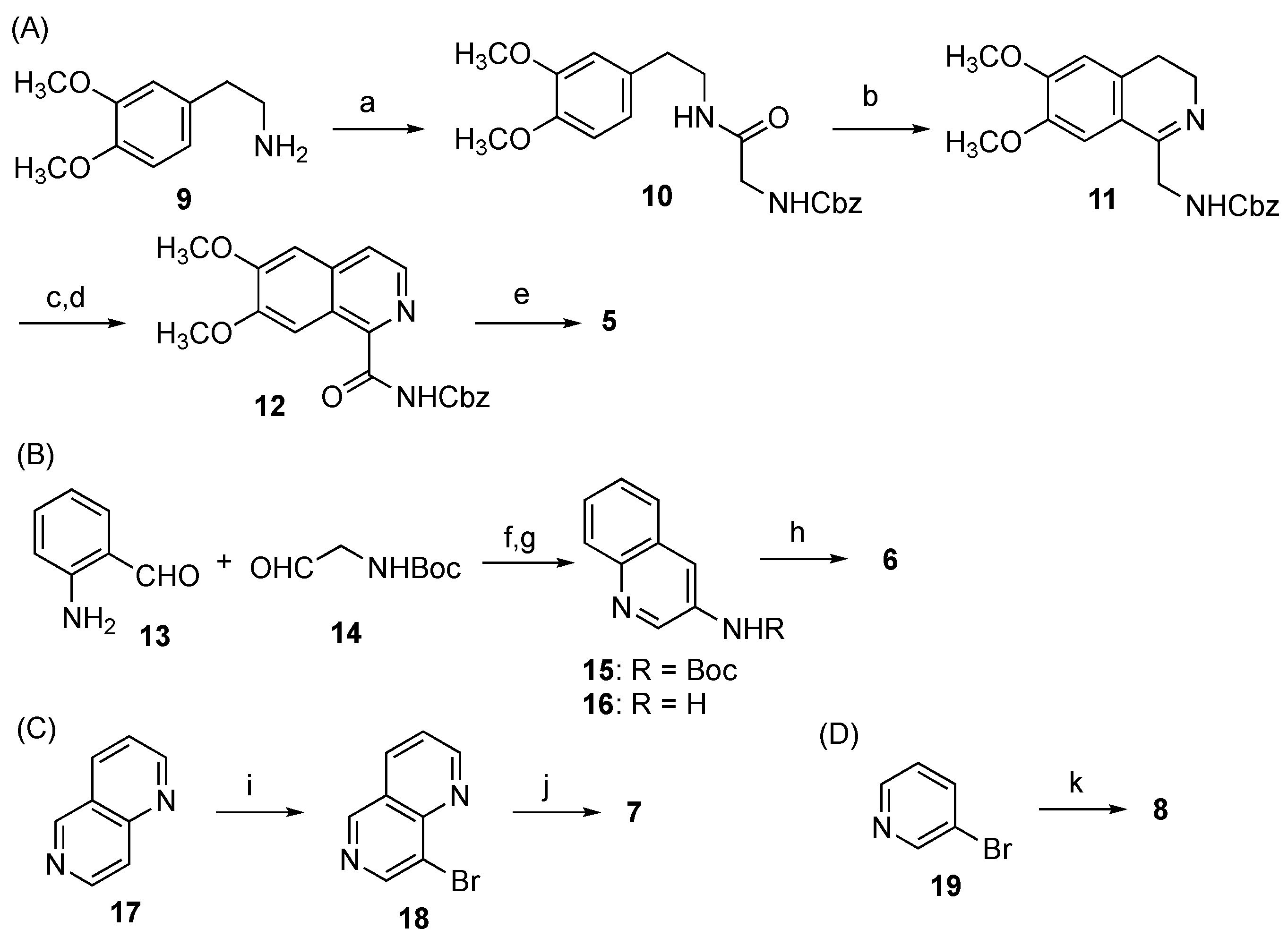
3.5.1. Benzyl (2-((3,4-Dimethoxyphenethyl)amino)-2-oxoethyl)carbamate (10)
EDCI·HCl (9.2 g, 48.1 mmol) and HOBt (3.8 g, 25.1 mmol) were added to a solution of homoveratrylamine (9, 4.6 g, 25.4 mmol) and Cbz-glycine (5.0 g, 23.9 mmol) in DMF (100 mL) and the whole mixture was stirred at rt for 2 h. AcOEt (30 mL) and 1 N HCl aq. were added to the mixture at 0 °C and the whole mixture was extracted with AcOEt. The organic phase was successively washed with sat. NaHCO3 aq. and brine. Removal of the solvent from the organic phase under reduced pressure gave 10 (7.93 g, 89%).
All the spectral data were identical to the reported ones [
29].
3.5.2. Benzyl ((6,7-Dimethoxy-3,4-dihydroisoquinolin-1-yl)methyl)carbamate (11)
POCl3 (11.9 mL, 128 mmol) was added to a solution of 10 (7.93 g, 21.3 mmol) in CH2Cl2 (210 mL), preheated at 45 °C. The mixture was stirred with reflux for 27 h. 28% NH3 aq. was added to the mixture at 0 °C and the whole mixture was extracted with CH2Cl2. Removal of the solvent from the organic phase under reduced pressure gave crude 11 (4.07 g, 54%), which was almost pure and was used for the next reaction without further purification.
All the spectral data were identical to the reported ones [
29].
3.5.3. Benzyl (6,7-Dimethoxyisoquinoline-1-carbonyl)carbamate (12)
A solution of 11 (10.7 mg, 0.030 mmol) in CHCl3 (0.5 mL) was stirred for 3 days under air. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was used for the next reaction without further purification.
1H NMR (400 MHz, CDCl3) δ: 10.03 (1H, brs), 8.01 (1H, s), 7.63–7.31 (5H, m), 6.68 (1H, s), 5.25 (2H, s), 3.92 (3H, s), 3.91 (3H, s), 3.78 (1H, t, J = 7.8 Hz), 2.66 (2H, t, J = 7.8 Hz). 13C NMR (150 MHz, CDCl3) δ: 161.6, 156.5, 151.6, 150.5, 147.3, 135.1, 131.9, 128.62, 128.58, 118.3, 111.4, 109.8, 67.5, 56.0, 55.9, 47.1, 25.3. IR (KBr): 3020, 1782, 1479, 1216, 1045, 758, 669 cm−1. ESI MS: m/z 369 [M + H]+. HR-ESI MS: m/z 369.1450, calcd for C20H21N2O5. Found: 369.1461.
Activated carbon (20.5 mg, 100 wt%) was added to a solution of the above product (20.0 mg, 0.054 mmol) in xylene (2.0 mL), and the whole mixture was stirred under an O2 atmosphere at 120 °C for 10 h. After cooling to rt, the mixture was filtered through a Celite pad. Removal of the solvent from the filtrate under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (n-Hexane/AcOEt = 2:1) to give 12 (5.8 mg, 29%) as a yellow solid.
1H NMR (300 MHz, CDCl3) δ: 10.85 (1H, brs), 9.08 (1H, s), 8.33 (1H, d, J = 5.3 Hz), 7.73 (1H, d, J = 5.3 Hz), 7.56–7.33 (5H, m), 7.09 (1H, s), 5.31 (2H, s), 4.08 (3H, s), 4.04 (3H, s). 13C NMR (150 MHz, CDCl3) δ: 164.2, 153.3, 152.2, 151.0, 142.1, 139.1, 135.4, 135.3, 128.7, 128.5, 124.7, 124.3, 105.1, 104.6, 67.6, 56.4, 56.1. IR (KBr): 3020, 1777, 1471, 1216, 1050, 757, 669 cm−1. ESI MS: m/z 389 [M + Na]+. HR-ESI MS: m/z 389.1113, calcd for C20H18N2O5Na. Found: 389.1117.
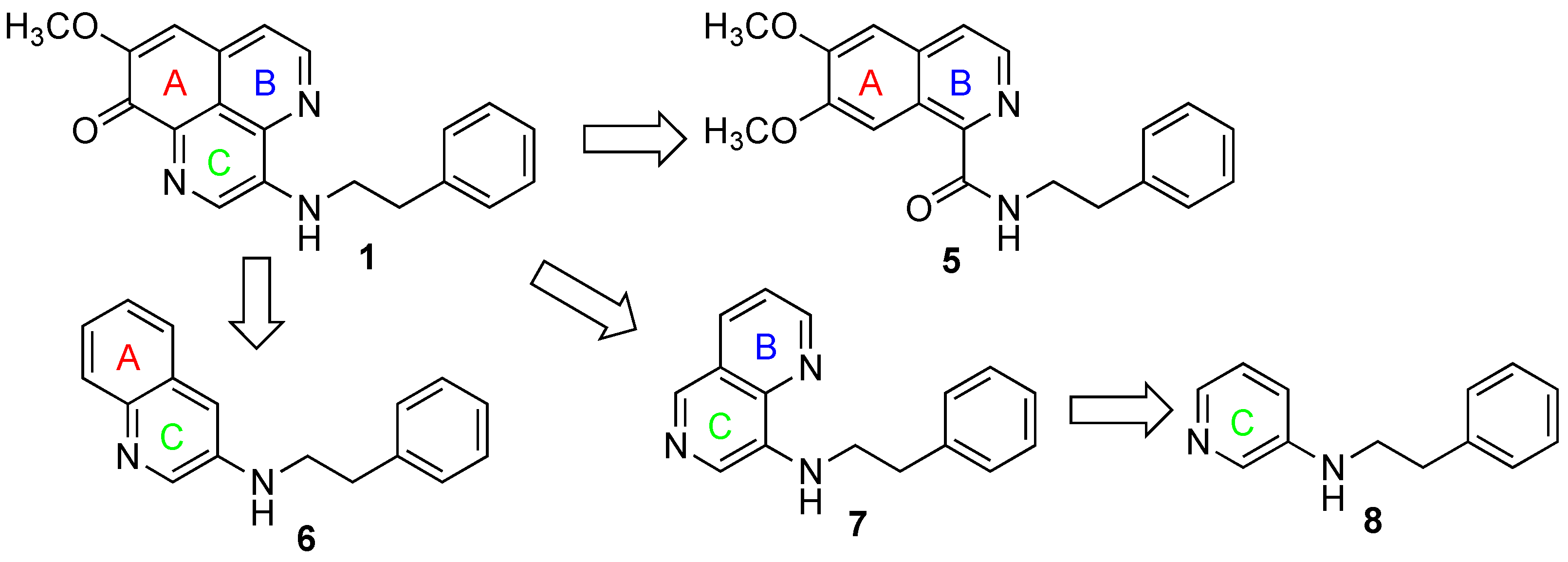
3.5.4. 6,7-Dimethoxy-N-phenethylisoquinoline-1-carboxamide (5)
NaH (5.2 mg, 0.11 mmol) was added to a solution of 12 (5.0 mg, 0.014 mmol) in DMF (0.5 mL) at 0 °C and the whole mixture was stirred for 5 min. Phenethyl bromide (20 µL, 0.16 mmol) was added to the mixture and the whole mixture was stirred for 24 h at rt, 48 at 60 °C, and 9 h at 90 °C. After cooling to rt, H2O (1 mL) was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with preparative TLC (n-Hexane/AcOEt = 2:1) to give 5 (3.3 mg, 72%) as a yellow solid.
1H NMR (400 MHz, CDCl3) δ: 9.36 (1H, s), 8.73 (1H, t-like), 8.49 (1H, d, J = 5.4 Hz), 7.83 (1H, d, J = 5.4 Hz), 7.65–7.40 (5H, m), 4.29 (3H, s), 4.23 (3H, s), 4.00–3.91 (2H, m), 3.19 (2H, t, J = 7.3 Hz). 13C NMR (150 MHz, CDCl3) δ: 166.7, 152.8, 151.1, 144.9, 139.1, 139.0, 134.9, 128.8, 128.6, 126.4, 122.9, 105.7, 104.4, 56.2, 56.0, 40.8, 36.0. IR (KBr): 3382, 2972, 1662, 1480, 1216, 760 cm−1. ESI MS: m/z 337 [M + H]+. HR-ESI MS: m/z 337.1552, calcd for C20H21N2O3. Found: 337.1544.
3.5.5. tert-Butyl quinolin-3-ylcarbamate (15)
4 N NaOH aq. (49 µL, 0.20 mmol) was added dropwise to a solution of 2-aminobenzaldehyde (13, 28.8 mg, 0.18 mmol) and tert-butyl (2-oxoethyl)carbamate (14, 7.9 mg, 0.065 mmol) in MeOH (0.5 mL) and the whole mixture was stirred at rt for 18 h. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was diluted with AcOEt and was then washed with H2O. Removal of the solvent from the AcOEt phase under reduced pressure gave a crude product, which was purified with preparative TLC (PTLC, CHCl3/MeOH = 60:1) to give 15 (10.4 mg, 52%) as a white solid.
All the spectral data were identical to the reported ones [
30].
3.5.6. Quinolin-3-amine (16)
TFA (120 µL) was added to a solution of 15 (6.4 mg, 0.026 mmol) in CH2Cl2 (1.0 mL) at 0 °C and the whole mixture was stirred at rt for 24 h. Sat. NaHCO3 aq. was added to the mixture and the whole mixture was extracted with CH2Cl2. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (CH2Cl2/MeOH = 80:1, 1% Et3N) to give 16 (3.6 mg, 95%)
All the spectral data were identical to the reported ones [
31].
3.5.7. N-Phenethylquinolin-3-amine (6)
6 was prepared through the reported method [
19]. All the spectral data were identical to the reported ones.
3.5.8. 8-Bromo-1,6-naphthyridine (18)
18 was prepared through the reported method [
32]. All the spectral data were identical to the reported ones.
3.5.9. N-Phenethyl-1,6-naphthyridin-8-amine (7)
Pd2(dba)3 (0.4 mg, 0.44 µmol) was added to a solution of rac-BINAP (0.6 mg, 0.96 µmol) in toluene (0.4 mL). After stirring at rt for 5 min, 18 (1.7 mg, 0.0081 mmol), 2-phenethylamine (1.1 µL, 0.0089 mmol) and t-BuONa (1.3 mg, 0.014 mmol) were successively added to the mixture and the whole mixture was stirred at 90 °C for 3 h. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified with PTLC (CHCl3/MeOH = 30:1) to give 7 (0.9 mg, 45%) as a tan solid.
1H NMR (500 MHz, CDCl3) δ: 8.88 (1H, dd, J = 4.3, 1.7 Hz), 8.59 (1H, s), 8.18 (1H, dd, J = 8.3, 1.7 Hz), 8.04 (1H, s), 7.49 (1H, dd, J = 8.3, 4.3 Hz), 7.39–7.29 (5H, m), 5.91 (1H, brs), 3.71–3.58 (2H, m), 3.09 (2H, t, J = 7.3 Hz). 13C NMR (150 MHz, CDCl3) δ 150.2, 138.1, 138.0, 134.4, 127.8, 127.6, 125.5, 123.6, 121.6, 43.6, 34.4. IR (KBr): 3020, 2927, 1216, 1028, 762 cm−1. MS (ESI-TOF) m/z: 250 [M + H]+. HRMS (ESI-TOF) m/z: 250.1344, calcd for C16H16N3. Found: 250.1344.
3.5.10. N-Phenethylpyridin-3-amine (8)
The flask containing BrettPhos/BrettPhos precatalyst (1:1, 13.2 mg, 0.02 mmol) and K2CO3 (331 mg, 2.4 mmol) was evacuated and was filled by Ar. 1,4-Dioxane (2.0 mL) was added to the flask and the whole mixture was stirred at rt for 10 min. 3-Bromopyridine (19, 96 µL, 1.0 mmol) and 2-phenethylamine (0.15 mL, 1.2 mmol) were then added to the mixture, and the whole mixture was stirred at reflux (oil bath temp. 110 °C) for 24 h. H2O was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (n-Hexane/AcOEt = 1:1) to give 8 (148 mg, 71%) as a colorless solid.
All the spectral data were identical to the reported ones [
21].
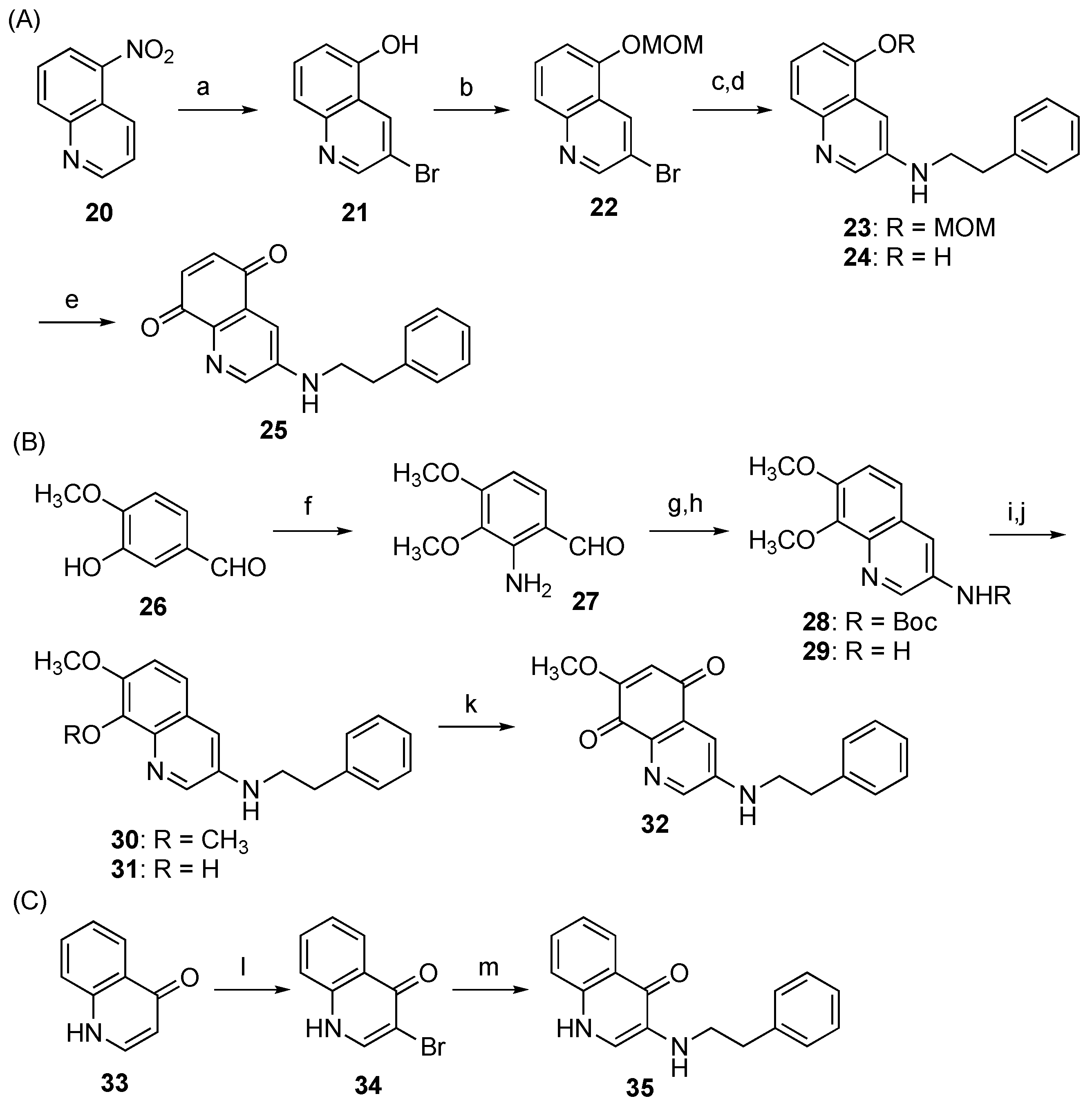
3.5.11. 3-Bromoquinolin-5-ol (21)
21 was prepared from commercially available 5-nitroquinoline (
20) through the reported method [
23]. All the spectral data were identical to the reported ones.
3.5.12. 3-Bromo-5-(methoxymethoxy)quinoline (22)
Chloromethyl methyl ether (84 μL, 1.10 mmol) and K2CO3 (408 mg, 2.95 mmol) were added to a solution of 21 (225 mg, 1.00 mmol) in acetone (5 mL) and the whole mixture was stirred at rt for 2 h. H2O was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt phase under reduced pressure gave a crude product containing 22, which was used for the next reaction without further purification.
3.5.13. 5-(Methoxymethoxy)-N-phenethylquinolin-3-amine (23)
An aliquot of 22 (53.6 mg, 0.20 mmol), 2-phenethylamine (63 μL, 0.399 mmol), Pd2(dba)3 (19.2 mg, 21.0 μmol), rac-BINAP (23.5 mg, 37.7 μmol), and t-BuONa (43.5 mg, 0.453 mmol) were dissolved in toluene (2 mL) and the whole mixture was stirred at 80 °C for 17 h. After cooling to rt, the reaction mixture was filtered through a Celite pad. The filtrate was concentrated under reduced pressure to give a crude product, which was used for the next reaction without further purification.
3.5.14. 3-(Phenethylamino)quinolin-5-ol (24)
Conc. HCl aq. (0.3 mL) was added to a solution of 23 (49.2 mg, 0.160 mmol) in MeOH (0.9 mL) and the whole mixture was stirred at rt for 3 h. The reaction mixture was neutralized with sat. NaHCO3 aq. And the whole mixture was extracted with CHCl3 containing 10% MeOH. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (CHCl3/MeOH = 10:1) to give 24 (29.9 mg, 70% in 3 steps) as a yellow solid.
1H NMR (500 MHz, CDCl3) δ: 9.30 (brs, 1H), 8.39 (d, J = 2.9 Hz, 1H), 7.61 (d, J = 2.7 Hz, 1H), 7.59 (d, J = 8.5 Hz, 1H), 7.33 (t, J = 7.3 Hz, 2H), 7.26–7.23 (m, 3H), 7.20 (d, J = 8.0 Hz, 1H), 6.86 (dd, J = 7.6, 0.6 Hz, 1H), 3.99 (brs, 1H), 3.52 (t, J = 6.9 Hz, 2H), 2.99 (t, J = 6.9 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 151.4, 142.6, 142.4, 140.8, 138.8, 128.8 (2C), 128.7 (2C), 126.6, 125.2, 121.5, 119.8, 109.5, 106.9, 44.7, 34.9. IR (KBr): 3413, 3019, 1608, 1476 cm–1. ESI MS: m/z 265 (M + H)+. HR-ESI MS: m/z 265.1341, calcd for C17H17N2O. Found: 265.1342.
3.5.15. 3-(Phenethylamino)quinoline-5,8-dione (25)
Fremy’s salt (60%, 76.2 mg, ca. 0.170 mmol) was dissolved to a solution of KH2PO4 (204 mg, 1.50 mmol) in H2O (30 mL), and a solution of 24 (15.0 mg, 56.7 μmol) in acetone (8 mL) was added dropwise to the mixture. After stirring the whole mixture at rt for 1 h, acetone was removed from the mixture under reduced pressure, and the resulting aqueous phase was extracted with CH2Cl2. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with PTLC (CHCl3/MeOH = 50:1) to give 25 (4.6 mg, 29%) as a red-purple solid.
1H NMR (600 MHz, CDCl3) δ: 8.28 (d, J = 2.9 Hz, 1H), 7.34 (t, J = 7.5 Hz, 2H), 7.32 (d, J = 2.9 Hz, 1H), 7.27 (t, J = 7.2 Hz, 1H), 7.22 (d, J = 7.3 Hz, 2H), 7.00 (d, J = 10.3 Hz, 1H), 6.91 (d, J = 10.3 Hz, 1H), 4.70 (brs, 1H), 3.59 (q, J = 6.5 Hz, 2H), 2.99 (t, J = 6.9 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ: 185.8, 182.2, 147.0, 140.9, 139.7, 137.8, 137.3, 137.0, 130.5, 128.9 (2C), 128.7 (2C), 127.0, 111.7, 44.1, 34.9. IR (KBr): 3619, 3020, 1672, 1579 cm–1. ESI MS: m/z 279 (M + H)+. HR-ESI MS: m/z 279.1134, calcd for C17H15N2O2. Found: 279.1127.
3.5.16. N-Phenethylquinolin-3-amine (27)
27 was prepared from isovanillin (
26) through the reported method [
24]. All the spectral data were identical to the reported ones.
3.5.17. tert-Butyl (7,8-dimethoxyquinolin-3-yl)carbamate (28)
4 N NaOH aq. (124 µL, 2.4 mmol) was added dropwise to a solution of 27 (25.1 mg, 0.21 mmol) and 14 (149 mg, 0.93 mmol) in MeOH (1.0 mL), and the whole mixture was stirred at rt for 30 h. MeOH was removed from the mixture under reduced pressure, and the resulting aqueous phase was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (n-hexane/AcOEt = 1:1) to give 28 (19.2 mg, 30%) as a tan oil.
1H NMR (300 MHz, CDCl3) δ: 8.63 (1H, s), 8.50 (1H, br), 7.49 (1H, d, J = 9.0 Hz), 7.32 (1H, d, J = 9.0 Hz), 7.08 (1H, s), 4.07 (3H, s), 3.99 (3H, s), 1.52 (9H, s). 13C NMR (151 MHz, CDCl3) δ 153.1, 150.3, 143.8, 143.0, 139.4, 130.9, 124.4, 122.9, 122.0, 116.2, 61.7, 56.9, 28.3. IR (KBr): 3433, 3020, 2401, 1712, 1525, 1370, 1216, 758 cm−1. ESI MS: m/z 327 [M + Na]+. HR-ESI MS: m/z 327.1321, calcd for C16H20N2O4Na. Found: 327.1305.
3.5.18. 7,8-Dimethoxyquinolin-3-amine (29)
TFA (0.17 mL, 2.2 mmol) was added to a solution of 28 (34.8 mg, 0.11 mmol) in CH2Cl2 (1.0 mL) at 0 °C, and the whole mixture was stirred at rt for 3 h. Sat. NaHCO3 aq. was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with PTLC (CHCl3/MeOH = 30:1) to give 29 (14.0 mg, 60%) as a tan oil.
1H NMR (500 MHz, CDCl3) δ: 8.54 (1H, d, J = 2.7 Hz), 7.32 (1H, d, J = 9.1 Hz), 7.27 (1H, d, J = 9.1 Hz), 7.21 (1H, d, J = 2.7 Hz), 4.10 (3H, s), 3.98 (3H, s), 3.83 (2H, brs). 13C NMR (151 MHz, CDCl3) δ 148.3, 143.7, 143.4, 138.5, 137.8, 125.4, 121.1, 116.5, 115.3, 61.8, 57.2. IR (KBr): 3394, 3019, 2400, 1626, 1484, 1347, 1216, 1109, 768 cm−1. MS (ESI-TOF) m/z: 205 [M + H]+. HRMS (ESI-TOF) m/z: 205.0977, calcd for C11H13N2O2. Found: 205.0986.
3.5.19. 7,8-Dimethoxy-N-phenethylquinolin-3-amine (30)
Pyridine (6.3 µL, 0.078 mmol) and Cu(OAc)2 (6.1 mg, 0.034 mmol) were added to a solution of 29 (5.3 mg, 0.026 mmol) in 1,4-dioxane (2.0 mL) and the whole mixture was stirred under reflux for 15 min. 2-Phenethylboronic acid (5.1 mg, 0.034 mmol) was added to the mixture and the whole mixture was further stirred under reflux for 14 h. After cooling to rt, H2O was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with PTLC (CHCl3/MeOH = 30:1) to give 30 (3.0 mg, 38%) as a red-purple solid.
1H NMR (500 MHz, CDCl3) δ: 8.42 (1H, d, J = 2.8 Hz), 7.36–7.32 (3H, m), 7.28–7.20 (4H, m), 7.02 (1H, d, J = 2.8 Hz), 4.10 (3H, s), 3.98 (3H, s), 3.91 (1H, t, J = 5.5 Hz), 3.49 (2H, dd, J = 12.9, 6.8 Hz), 3.00 (2H, t, J = 7.0 Hz). 13C NMR (150 MHz, CDCl3) δ 148.5, 143.9, 143.7, 140.1, 138.8, 137.2, 128.8, 126.7, 125.7, 121.1, 116.5, 110.7, 61.8, 57.3, 44.8, 35.1. IR (KBr): 3413, 3020, 2400, 1610, 1511, 1382, 1216, 773 cm−1. MS (ESI-TOF) m/z: 309 [M + H]+. HRMS (ESI-TOF) m/z: 309.1603, calcd for C19H21N2O2. Found: 309.1618.
3.5.20. 7-Methoxy-3-(phenethylamino)quinolin-8-ol (31)
A solution of 30 (30.5 mg, 98.9 μmol) in 48% HBr aq. (2.5 mL) was stirred at 100 °C for 3 h. Sat. NaHCO3 aq. was added to the mixture and the whole mixture was extracted with CH2Cl2. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with PTLC (CHCl3/MeOH = 20:1) to give 31 (19.8 mg, 68%) as a red-purple solid.
1H NMR (500 MHz, CDCl3) δ: 8.28 (s, 1H), 7.37 (t, J = 7.2 Hz, 2H), 7.31–7.24 (m, 4H), 7.13 (d, J = 9.2 Hz, 1H), 7.06 (s, 1H), 4.02 (s, 3H), 3.51 (t, J = 6.3 Hz, 2H), 3.02 (t, J = 6.3 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 142.8, 141.8, 141.1, 140.7, 138.8, 132.7, 128.8 (4C), 126.7, 124.8, 117.4, 115.5, 110.9, 57.6, 44.8, 35.0. IR (KBr): 3154, 2932, 2253, 1791, 1609, 1469, 1383 cm−1. MS (ESI-TOF) m/z: 295 [M + H]+. HRMS (ESI-TOF) m/z: 295.1441, calcd for C18H19N2O2. Found: 295.1452.
3.5.21. 7-Methoxy-3-(phenethylamino)quinoline-5,8-dione (32)
Using the same synthetic procedure as that of 25, 31 (12.0 mg, 40.7 μmol) was converted to 32 (6.0 mg, 47%) as a red-purple solid.
1H NMR (600 MHz, CDCl3) δ 8.24 (d, J = 2.7 Hz, 1H), 7.35 (t, J = 7.0 Hz, 3H), 7.28 (d, J = 7.3 Hz, 1H), 7.22 (d, J = 7.5 Hz, 2H), 6.11 (s, 1H), 4.64 (s, 1H), 3.91 (s, 3H), 3.60 (q, J = 6.5 Hz, 2H), 2.99 (t, J = 6.8 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ: 184.7, 176.9, 161.5, 147.3, 140.4, 137.7, 136.4, 130.9, 128.9 (4C), 128.7, 127.0, 111.7, 108.4, 56.6, 44.0, 34.9. IR (KBr): 2253, 1672, 1646, 1579, 1260, 1231, 1073 cm−1. ESI MS: m/z 331 (M + Na)+. HR-ESI MS: m/z 331.1059, calcd for C18H16N2O3Na. Found: 331.1047.
3.5.22. 3-Bromoquinolin-4(1H)-one (34)
Bromine (52 μL, 1.00 mmol) was added to a solution of quinolin-4(1H)-one (33, 147 mg, 1.01 mmol) in AcOH (2 mL) and the whole mixture was stirred at reflux (oil bath temp. 120 °C) for 2 h. After cooling to rt, ice water (8 mL) and 1 N Na2S2O3 aq. (2 mL) were added to the mixture, and the whole mixture was vigorously stirred for 15 min. Suction filtration of the precipitated white solid gave 34 (198 mg, 88%).
All the spectral data were identical to the reported ones [
33].
3.5.23. 3-(Phenethylamino)quinolin-4(1H)-one (35)
CuSO4 (0.3 mg, 1.88 μmol) was added to a solution of 34 (44.8 mg, 0.200 mmol) in 2-phenethylamine (200 μL, 1.71 mmol) and the whole mixture was stirred at 150 °C for 56 h. H2O was added to the mixture and the whole mixture was extracted with CH2Cl2. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (hexane/AcOEt = 1:1 then CHCl3/MeOH = 10:1) to give 35 (34.3 mg, 27%) as a yellow solid.
1H NMR (600 MHz, CDCl3) δ: 11.66 (brs, 1H), 8.38 (d, J = 8.3 Hz, 1H), 7.53 (d, J = 8.5 Hz, 1H), 7.43 (td, J = 8.4, 1.5 Hz, 1H), 7.33 (d, J = 5.2 Hz, 1H), 7.22–7.19 (m, 3H), 7.17–7.12 (m, 3H), 4.66 (brs, 1H), 3.30 (t, J = 7.2 Hz, 2H), 2.94 (t, J = 7.2 Hz, 2H).13C NMR (150 MHz, CDCl3) δ: 170.4, 139.1, 137.5, 133.1, 130.1, 128.6 (2C), 128.5 (2C), 126.4, 125.0, 122.2, 121.6, 118.4, 117.5, 46.8, 35.5. IR (KBr): 3063, 2939, 1633, 1559, 1497, 1460, 754, 699 cm–1. ESI MS: m/z 265 (M + H)+. HR-ESI MS: m/z 265.1341, calcd for C17H17N2O. Found: 265.1341.
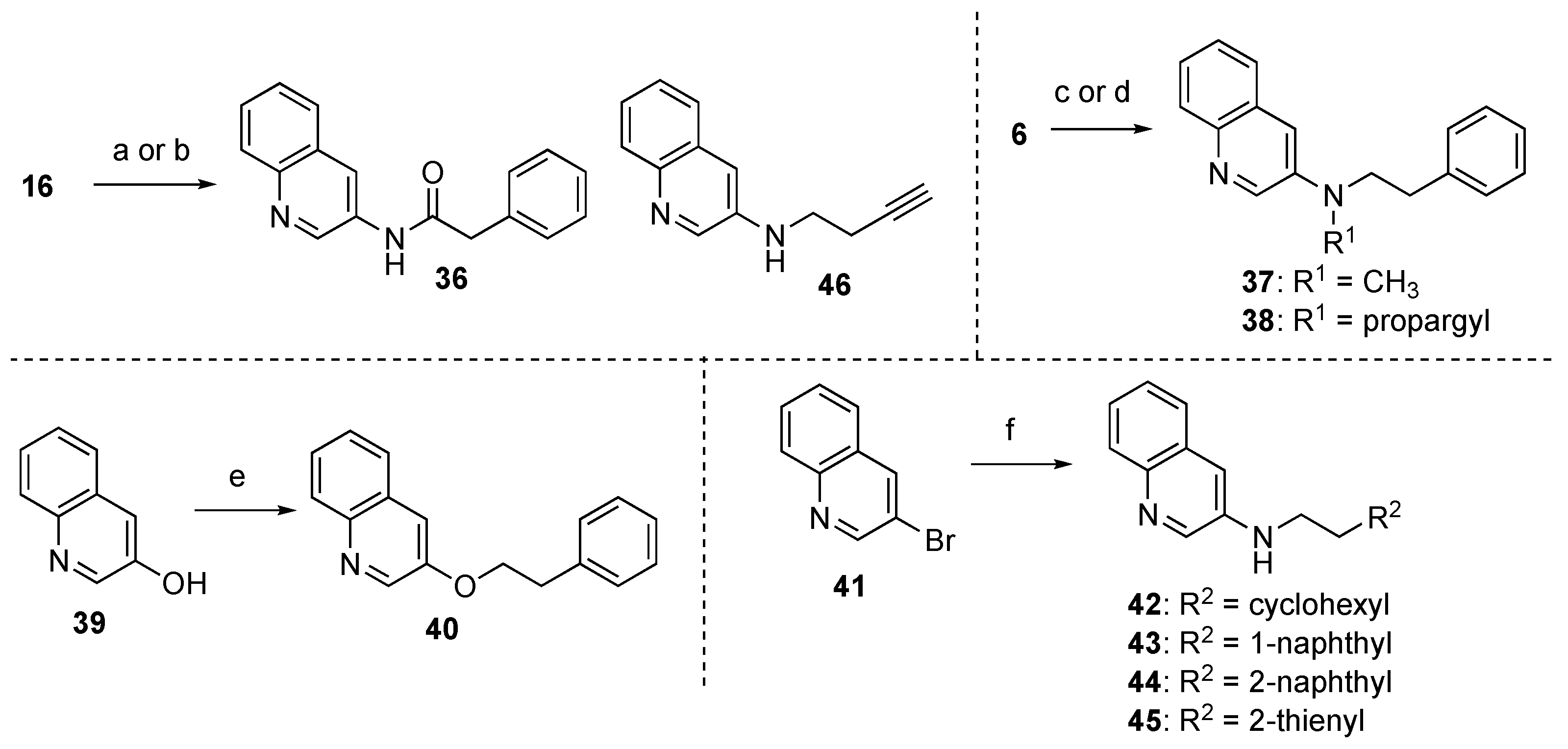
3.5.24. 2-Phenyl-N-(quinolin-3-yl)acetamide (36)
A solution of phenacyl chloride (93 μL, 0.704 mmol) in CH2Cl2 (2 mL) was added dropwise to a solution of 16 (68.4 mg, 0.474 mmol) and pyridine (402 μL, 4.99 mmol) in CH2Cl2 (3 mL) and the whole mixture was stirred at rt for 7 h. Sat. NH4Cl aq. was added to the mixture and the whole mixture was extracted with CH2Cl2. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (hexane/AcOEt = 1:1) to give 36 (82.5 mg, 66%) as a white solid.
1H NMR (600 MHz, CDCl3) δ: 8.71 (d, J = 2.5 Hz, 1H), 8.60 (d, J = 2.6 Hz, 1H), 8.18 (s, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.73 (d, J = 8.2 Hz, 1H), 7.59 (t, J = 7.6 Hz, 1H), 7.50 (t, J = 7.5 Hz, 1H), 7.37 (t, J = 7.4 Hz, 1H), 7.34–7.30 (m, 3H), 3.79 (s, 2H). 13C NMR (150 MHz, CDCl3) δ: 170.1, 145.0, 143.8, 134.0, 131.4, 129.4 (2C), 129.2 (2C), 128.6, 128.4, 128.1, 127.7 (2C), 127.3, 124.1, 44.5. IR (KBr): 3019, 1689, 1530 cm−1. ESI MS: m/z 263 (M + H)+. HR-ESI MS: m/z 263.1179, calcd for C17H15N2O. Found: 263.1170.
3.5.25. N-Methyl-N-phenethylquinolin-3-amine (37)
A solution of 6 (25.0 mg, 0.101 mmol) in 2,2,2-trifluoroethanol (TFE, 0.25 mL) was added to a solution of HCHO aq. (18 μL, 0.500 mmol) in TFE (0.25 mL) and the whole mixture was stirred at rt for 5 min. NaBH4 (7.6 mg, 0.201 mmol) was added to the mixture and the whole mixture was stirred at rt for 13 h. The reaction was quenched by the addition of H2O, and the whole mixture was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (CHCl3/MeOH = 20:1) to give 37 (20.4 mg, 77%) as a pale yellow oil.
1H NMR (600 MHz, CDCl3) δ: 8.69 (d, J = 3.0 Hz, 1H), 7.96 (dd, J = 6.3, 2.6 Hz, 1H), 7.64 (dd, J = 7.5, 2.1 Hz, 1H), 7.45–7.40 (m, 2H), 7.31 (t, J = 7.5 Hz, 2H), 7.25–7.21 (m, 3H), 7.11 (d, J = 3.0 Hz, 1H), 3.73 (t, J = 7.5 Hz, 2H), 3.00 (s, 3H), 2.90 (t, J = 7.6 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ: 142.3, 141.2, 140.9, 139.1, 129.3, 128.8 (4C), 128.6, 126.8, 126.4, 126.0, 124.9, 112.2, 54.6, 38.5, 33.2. IR (KBr): 3019, 2957, 1599 cm−1. ESI MS: m/z 263 (M + H)+. HR-ESI MS: m/z 263.1543, calcd for C18H19N2. Found: 263.1550.
3.5.26. N-Phenethyl-N-(prop-2-yn-1-yl)quinolin-3-amine (38)
K2CO3 (2.2 mg, 15.9 μmol) and propargyl bromide (52 μL, 0.480 mmol) were added to a solution of 6 (40.0 mg, 0.161 mmol) in acetone (2.4 mL) and the whole mixture was stirred at 60 °C for 32 h. After cooling to rt, H2O was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (hexane/AcOEt = 2:1) to give 38 (7.0 mg, 15%) as a pale yellow oil.
1H NMR (500 MHz, CDCl3) δ: 8.71 (d, J = 2.9 Hz, 1H), 7.97 (dd, J = 6.3, 2.9 Hz, 1H), 7.69–7.67 (m, 1H), 7.49–7.44 (m, 2H), 7.34–7.31 (m, 3H), 7.25–7.23 (m, 2H), 4.09 (t, J = 2.3 Hz, 2H), 3.76 (t, J = 7.4 Hz, 2H), 3.01 (t, J = 7.4 Hz, 2H), 2.27 (t, J = 5.2 Hz, 1H). 13C NMR (125 MHz, CDCl3) δ: 142.2, 142.1, 141.3, 139.1, 129.1, 129.0, 128.9 (2C), 128.8 (2C), 127.0, 126.7, 126.4, 125.8, 114.8, 79.2, 73.0, 53.5, 40.5, 34.1. IR (KBr): 3155, 2253, 1217 cm−1. ESI MS: m/z 287 (M + H)+. HR-ESI MS: m/z 287.1543, calcd for C20H19N2. Found: 287.1539.
3.5.27. 3-Phenethoxyquinoline (40)
NaH (60.0 mg, ca. 1.50 mmol) and 2-phenethyl bromide (205 μL, 1.52 mmol) were added to a solution of quinolin-3-ol (39) (149 mg, 1.03 mmol) in DMF (2 mL) and the whole mixture was stirred at rt for 18 h. Sat. NaHCO3 aq. was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (hexane/EtOAc = 1:1) to give 40 (108 mg, 42%) as a pale yellow oil.
1H NMR (600 MHz, CDCl3) δ: 8.69 (d, J = 2.9 Hz, 1H), 8.05 (d, J = 8.4 Hz, 1H), 7.69 (dd, J = 8,2, 1.5 Hz, 1H), 7.55 (td, J = 8.3, 1.5 Hz, 1H), 7.50 (td, J = 8.2, 1.3 Hz, 1H), 7.37–7.32 (m, 5H), 7.28 (tt, J = 6.9, 1.9 Hz, 1H), 4.30 (t, J = 7.1 Hz, 2H), 3.20 (t, J = 7.1 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ: 152.2, 144.7, 143.4, 137.7, 129.1, 129.0 (2C), 128.7, 128.6 (2C), 127.0, 126.7, 126.6 (2C), 112.9, 68.9, 35.5. IR (KBr): 3019, 2953, 1604, 1346, 1216 cm−1. ESI MS: m/z 250 (M + H)+. HR-ESI MS: m/z 250.1232, calcd for C17H16NO. Found: 250.1241.
3.5.28. N-(2-Cyclohexylethyl)quinolin-3-amine (42)
With the same synthetic procedure as that of 7, 3-bromoquinoline (41, 61 μL, 0.454 mmol) was converted to 42 (102.1 mg, 88%) using 2-(cyclohexyl)ethylamine (72 μL, 0.500 mmol) as a colorless oil.
1H NMR (500 MHz, CDCl3) δ: 8.42 (d, J = 2.8 Hz, 1H), 7.92 (dd, J = 7.3, 1.6 Hz, 1H), 7.60 (dd, J = 7.8, 1.7 Hz, 1H), 7.45–7.35 (m, 2H), 6.98 (d, J = 2.8 Hz, 1H), 3.92 (s, 1H), 3.21 (td, J = 7.3, 4.3 Hz, 2H), 2.60 (d, J = 1.4 Hz, 1H), 1.82–1.63 (m, 3H), 1.63–1.54 (m, 2H), 1.49–1.37 (m, 1H), 1.33–1.11 (m, 3H), 0.98 (qd, J = 11.9, 3.1 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 143.4, 141.9, 141.8, 129.6, 129.0, 126.8, 125.8, 124.6, 109.6, 41.3, 36.7, 35.5, 33.3, 26.5, 26.2. IR (KBr): 3423, 3019, 2925, 2853, 1611 cm−1. ESI MS: m/z 255 (M + H)+. HR-ESI MS: m/z 255.1856, calcd for C17H23N2. Found: 255.1866.
3.5.29. N-(2-(Naphthalen-1-yl)ethyl)quinolin-3-amine (43)
With the same synthetic procedure as that of 7, 41 (9.4 μL, 70 µmol) was converted to 43 (13.5 mg, 64%) using 2-(naphthalen-1-yl)ethylamine (17.1 mg, 0.10 mmol) as a yellow solid.
1H NMR (600 MHz, CDCl3) δ: 8.37 (d, J = 2.8 Hz, 1H), 8.07 (d, J = 8.2 Hz, 1H), 7.95 (dd, J = 6.6, 2.5 Hz, 1H), 7.90 (dd, J = 7.8, 1.6 Hz, 1H), 7.79 (d, J = 8.2 Hz, 1H), 7.61–7.59 (m, 1H), 7.56–7.51 (m, 2H), 7.45–7.40 (m, 3H), 7.38 (d, J = 6.9 Hz, 1H), 7.05 (d, J = 8.2 Hz, 1H), 4.06 (brs, 1H), 3.65 (q, J = 6.5 Hz, 2H), 3.47 (t, J = 6.9 Hz, 2H). 13C NMR (150 MHz, CDCl3) δ: 143.4, 142.1, 141.2, 134.8, 134.0, 131.8, 129.4, 129.0 (2C), 127.5, 126.9, 126.8, 126.2, 125.9, 125.8, 125.5, 124.9, 123.3, 110.2, 43.9, 32.1. IR (KBr): 3049, 1610, 1510, 1390, 1220, 778 cm−1. ESI MS: m/z 299 (M + H)+. HR-ESI MS: m/z 299.1548, calcd for C21H19N2. Found: 299.1537.
3.5.30. N-(2-(Naphthalen-2-yl)ethyl)quinolin-3-amine (44)
With the same synthetic procedure as that of 7, 41 (46 μL, 0.35 mmol) was converted to 44 (97.5 mg, 92%) using 2-(naphthalen-2-yl)ethylamine (66.4 mg, 0.39 mmol) as a yellow solid.
1H NMR (500 MHz, CDCl3) δ 8.39 (d, J = 2.8 Hz, 1H), 7.97–7.91 (m, 1H), 7.87–7.78 (m, 3H), 7.72–7.68 (m, 1H), 7.67–7.59 (m, 1H), 7.53–7.36 (m, 5H), 7.10 (d, J = 2.8 Hz, 1H), 4.03 (brs, 1H), 3.61 (t, J = 6.9 Hz, 2H), 3.18 (t, J = 6.9 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ 143.4, 142.1, 141.2, 136.2, 133.6, 132.3, 129.5, 128.9, 128.5, 127.7, 127.5, 127.2, 127.0 (2C), 126.3, 125.9, 125.7, 125.0, 110.4, 44.5, 35.1. IR (KBr): 3413, 3019, 1611, 1516, 1030 cm−1. ESI MS: m/z 299 (M + H)+. HR-ESI MS: m/z 299.1548, calcd for C21H19N2. Found: 299.1548.
3.5.31. N-(2-(Thiophen-2-yl)ethyl)quinolin-3-amine (45)
With the same synthetic procedure as that of 7, 41 (67 μL, 0.50 mmol) was converted to 45 (114 mg, 90%) using 2-(thiophen-2-yl)ethylamine (117 mg, 1.0 mmol) as a yellow solid.
1H NMR (500 MHz, CDCl3) δ 8.38 (d, J = 2.9 Hz, 1H), 7.99–7.92 (m, 1H), 7.66–7.58 (m, 1H), 7.49–7.37 (m, 2H), 7.18 (dd, J = 5.1, 1.2 Hz, 1H), 7.03 (d, J = 2.8 Hz, 1H), 6.97 (dd, J = 5.1, 3.4 Hz, 1H), 6.88 (dd, J = 3.4, 1.1 Hz, 1H), 4.25 (brs, 1H), 3.50 (t, J = 8.3 Hz, 2H), 3.19 (t, J = 6.7 Hz, 2H). 13C NMR (125 MHz, CDCl3) δ: 143.3, 142.0, 141.1, 141.0, 129.4, 128.8, 127.0, 126.9, 125.8, 125.4, 124.9, 124.0, 110.2, 44.7, 29.1. IR (KBr): 3405, 3256, 3054, 2927, 2849, 1613, 1517, 1222, 700 cm−1. ESI MS: m/z 255 (M + H)+. HR-ESI MS: m/z 255.0956, calcd for C15H15N2S. Found: 255.0946.
3.5.32. N-(But-3-yn-1-yl)quinolin-3-amine (46)
4-Bromobut-1-yne (92 μL, 0.997 mmol) was added to a solution of 16 (145 mg, 1.00 mmol) K2CO3 (153 mg, 1.11 mmol) in DMF (6 mL) and the whole mixture was stirred at 85 °C for 11 h. After cooling to rt, H2O was added to the mixture and the whole mixture was extracted with AcOEt. Removal of the solvent from the organic phase under reduced pressure gave a crude product, which was purified with SiO2 column chromatography (hexane/AcOEt = 1:1) to give 46 (14.8 mg, 7%) as a yellow solid.
1H NMR (600 MHz, CDCl3) δ: 8.47 (d, J = 2.9 Hz, 1H), 7.95–7.93 (m, 1H), 7.64–7.61 (m, 1H), 7.45–7.41 (m, 2H), 7.05 (d, J = 2.8 Hz, 1H), 4.28 (brs, 1H), 3.43 (q, J = 6.3 Hz, 2H), 2.60 (td, J = 6.6, 2.7 Hz, 2H), 2.09 (t, J = 2.6 Hz, 1H). 13C NMR (150 MHz, CDCl3) δ: 143.4, 142.3, 140.9, 129.3, 129.0, 127.0, 125.9, 125.1, 110.5, 81.2, 70.5, 42.0, 18.8. IR (KBr): 3409, 3307, 3154, 3056, 1793, 1614, 1515, 1483 cm−1. ESI MS: m/z 197 (M + H)+. HR-ESI MS: m/z 197.1079, calcd for C13H13N2. Found: 197.1070.


 and
and 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




