
N-[1,3-Dialkyl(aryl)-2-oxoimidazolidin-4-ylidene]-aryl(alkyl)sulphonamides as Novel Selective Human Cannabinoid Type 2 Receptor (hCB2R) Ligands; Insights into the Mechanism of Receptor Activation/Deactivation
 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
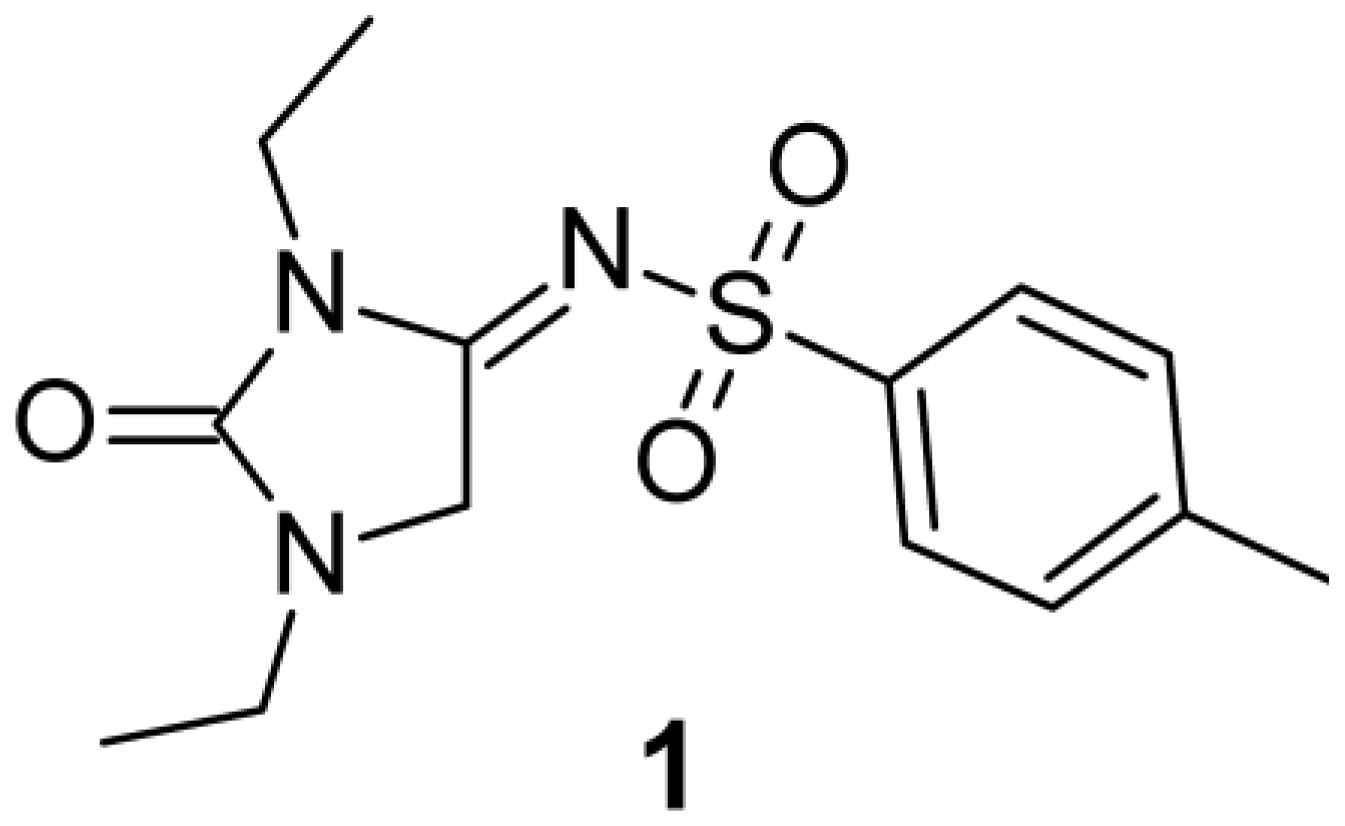
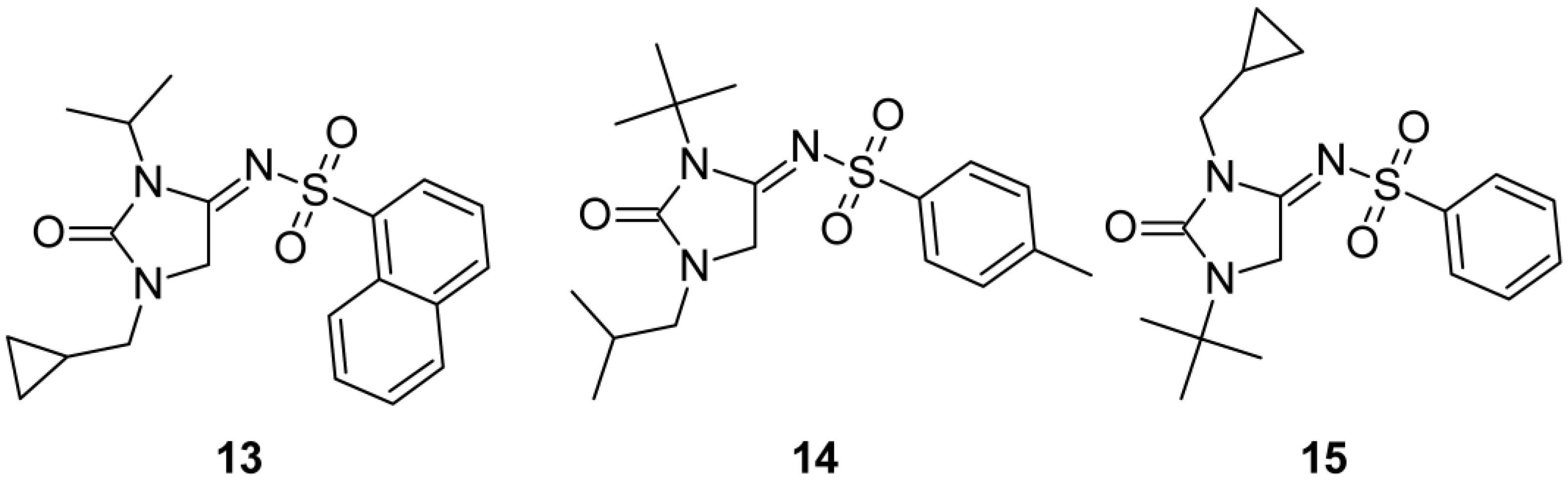
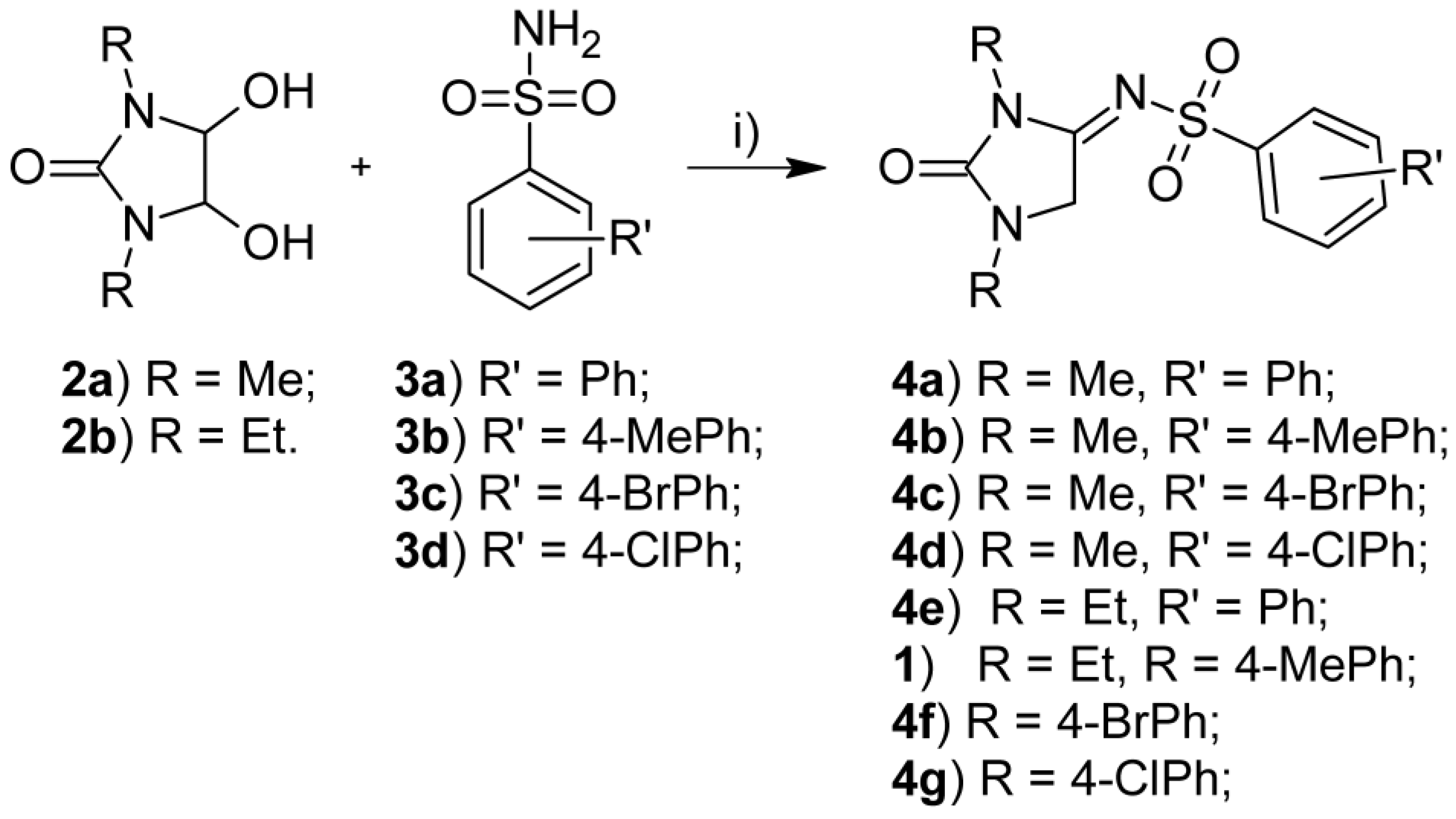
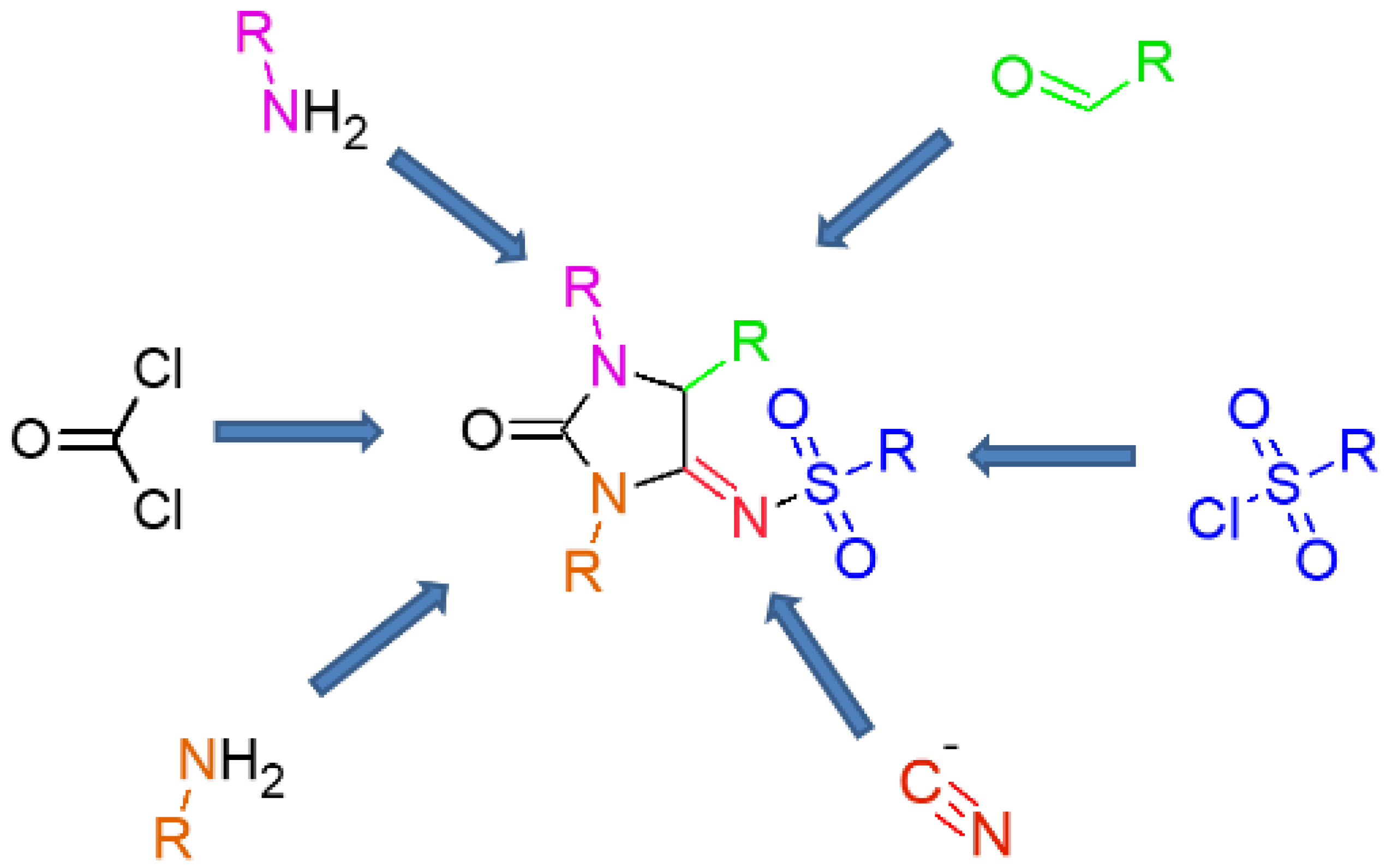
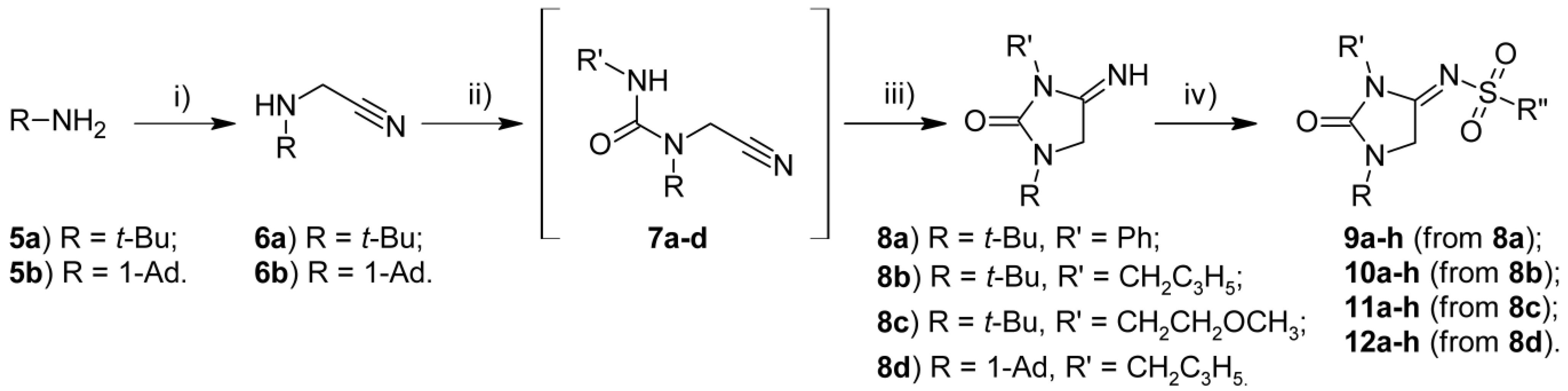
2.1. Chemistry
2.2. Competition Binding Assay
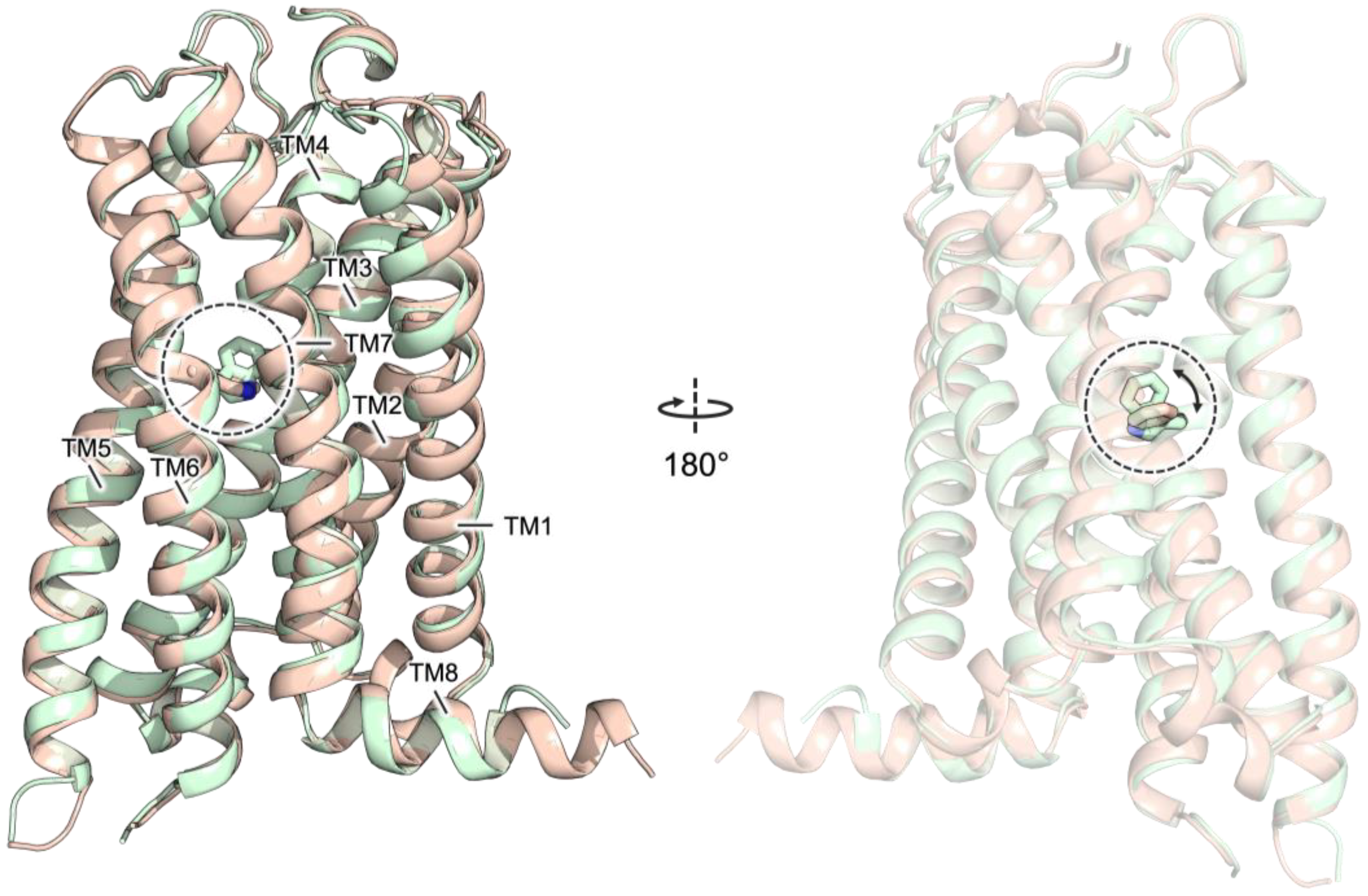
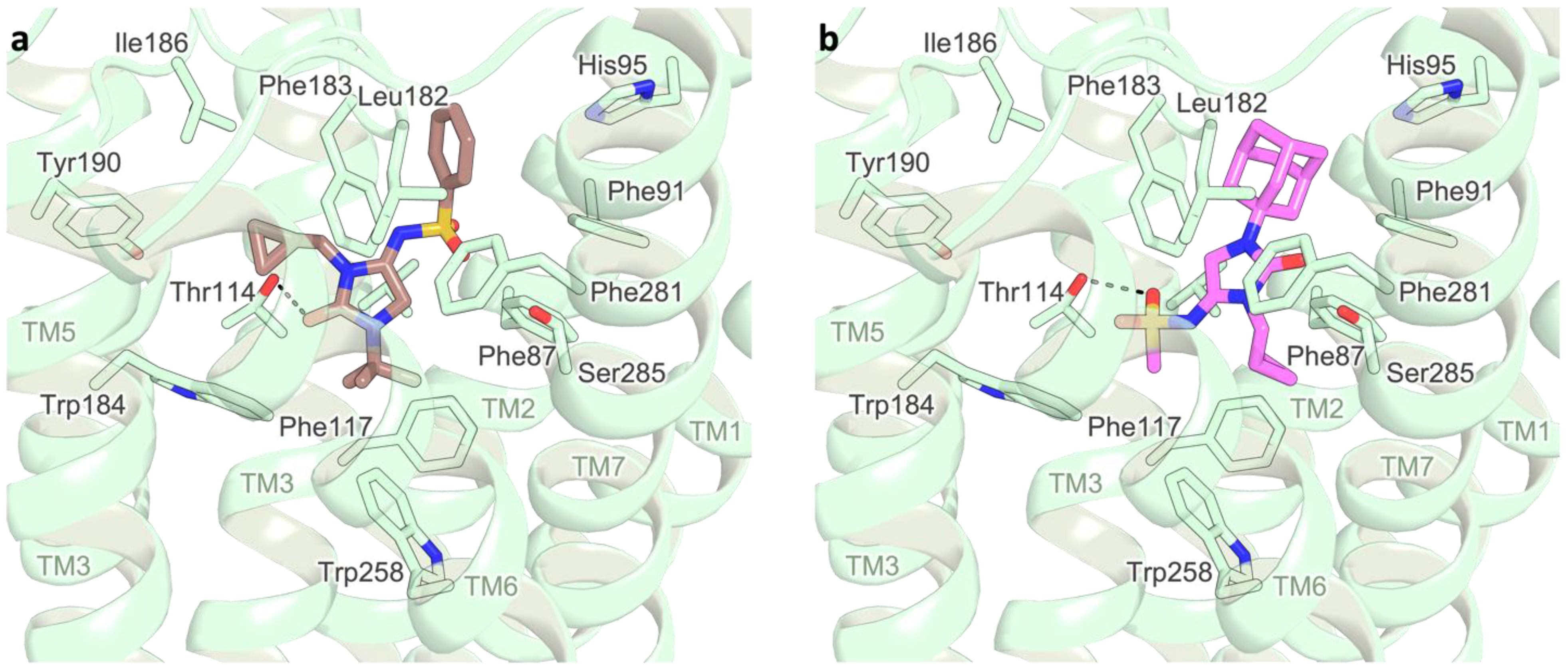
2.3. Docking Studies
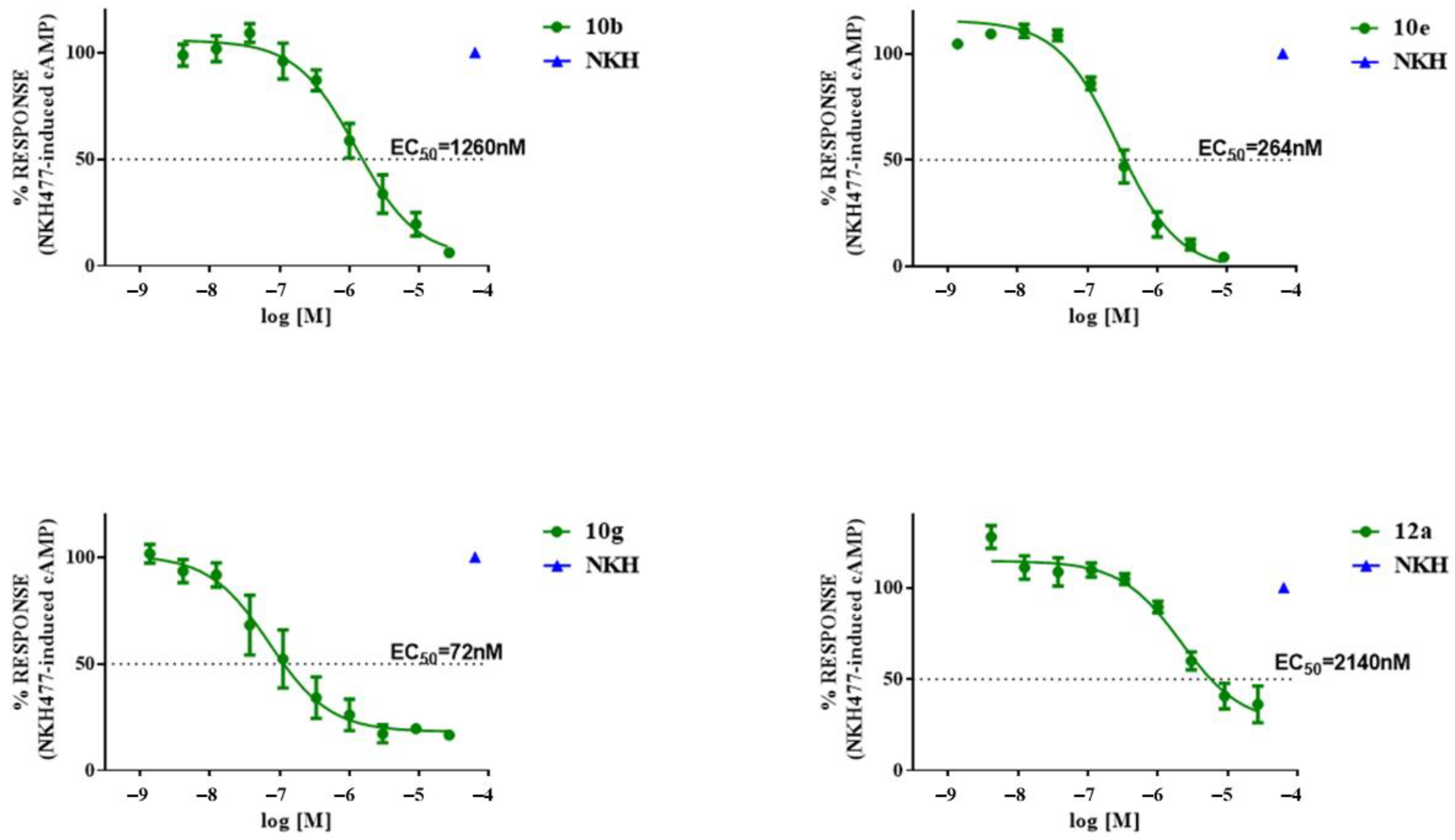
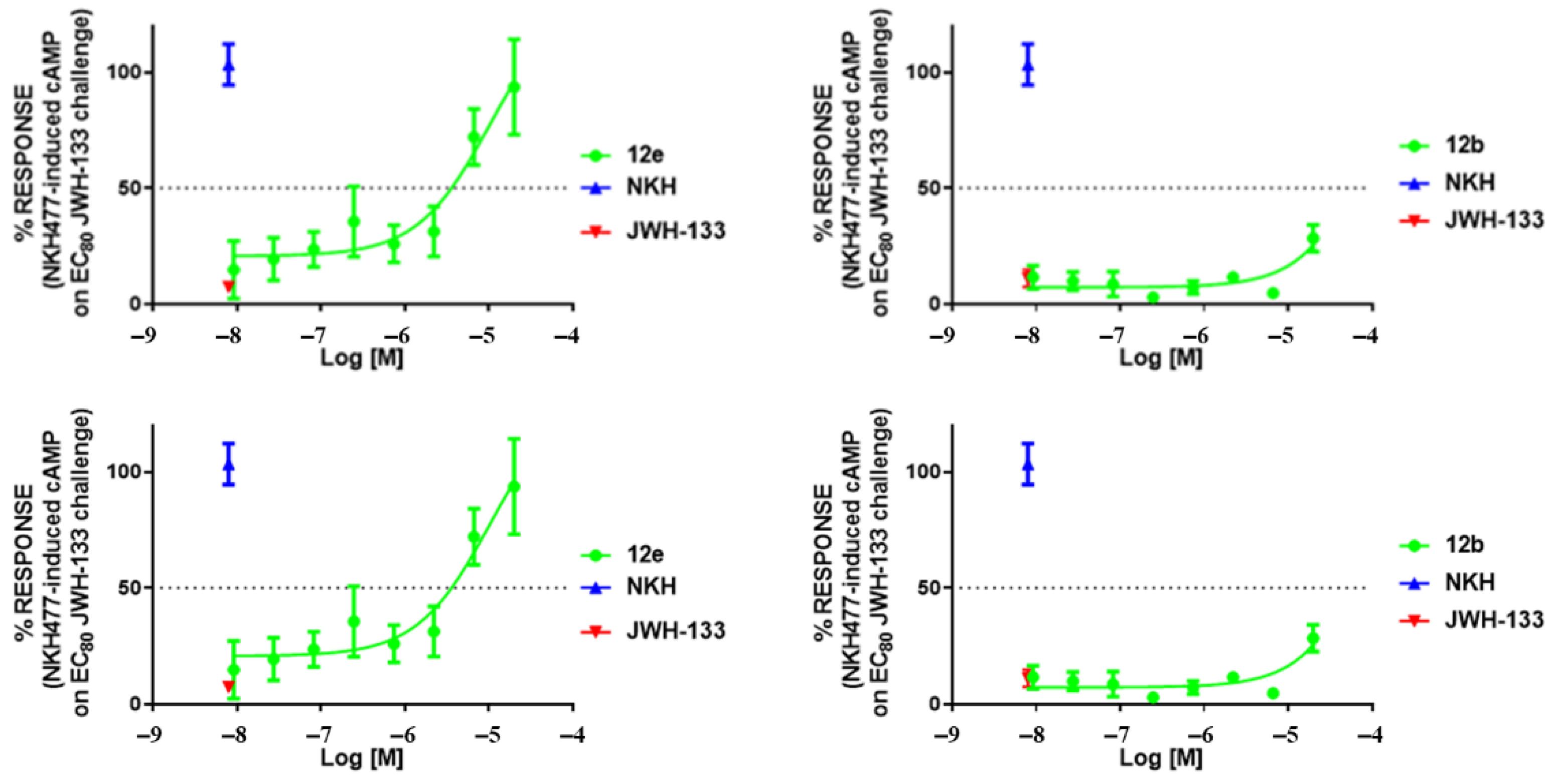
2.4. Functional Activity
3. Materials and Methods
3.1. Chemical Synthesis
3.1.1. General Synthetic Procedure for N-[1,3-Dialkyl-2-oxoimidazolidin-4-ylidene]arylsulfonamides
3.1.2. General Synthetic Procedure for N-[1-tert-Butyl-2-oxo-3-phenylimidazolidin-4-ylidene]sulphonamides (9a–h)
3.1.3. General Synthetic Procedure for N-[2-Oxoimidazolidin-4-ylidene]sulphonamides (10a–h–12a–h)
3.2. Molecular Docking Simulations
3.3. In Vitro Pharmacological Evaluation
3.3.1. Competition Binding Assay
3.3.2. Functional Activity at CB2R In Vitro
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bridgeman, M.B.; Abazia, D.T. Medicinal Cannabis: History, Pharmacology, And Implications for the Acute Care Setting. Pharm. Ther. 2017, 42, 180–188. [Google Scholar] [PubMed]
- Kirkham, T.C. Endocannabinoids in the Regulation of Appetite and Body Weight. Behav. Pharmacol. 2005, 16, 297–313. [Google Scholar] [CrossRef] [PubMed]
- Seeley, R.J.; Woods, S.C. Monitoring of Stored and Available Fuel by the CNS: Implications for Obesity. Nat. Rev. Neurosci. 2003, 4, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Calignano, A.; La Rana, G.; Giuffrida, A.; Piomelli, D. Control of Pain Initiation by Endogenous Cannabinoids. Nature 1998, 394, 277–281. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Stella, N. Cannabinoids and Neuroinflammation. Br. J. Pharmacol. 2004, 141, 775–785. [Google Scholar] [CrossRef] [Green Version]
- Fulmer, M.L.; Thewke, D.P. The Endocannabinoid System and Heart Disease: The Role of Cannabinoid Receptor Type 2. Cardiovasc. Hematol. Disord. Drug Targets 2018, 18, 34–51. [Google Scholar] [CrossRef]
- Idris, A.I.; Ralston, S.H. Role of Cannabinoids in the Regulation of Bone Remodeling. Front. Endocrinol. 2012, 3, 136. [Google Scholar] [CrossRef] [Green Version]
- Śledziński, P.; Zeyland, J.; Słomski, R.; Nowak, A. The Current State and Future Perspectives of Cannabinoids in Cancer Biology. Cancer Med. 2018, 7, 765–775. [Google Scholar] [CrossRef]
- Gaoni, Y.; Mechoulam, R. Isolation, Structure, and Partial Synthesis of an Active Constituent of Hashish. J. Am. Chem. Soc. 1964, 86, 1646–1647. [Google Scholar] [CrossRef]
- Devane, W.A.; Dysarz, F.A.; Johnson, M.R.; Melvin, L.S.; Howlett, A.C. Determination and Characterization of a Cannabinoid Receptor in Rat Brain. Mol. Pharmacol. 1988, 34, 605. [Google Scholar] [PubMed]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an Endogenous 2-Monoglyceride, Present in Canine Gut, That Binds to Cannabinoid Receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, T.; Kondo, S.; Sukagawa, A.; Nakane, S.; Shinoda, A.; Itoh, K.; Yamashita, A.; Waku, K. 2-Arachidonoylgylcerol: A Possible Endogenous Cannabinoid Receptor Ligand in Brain. Biochem. Biophys. Res. Commun. 1995, 215, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular Characterization of a Peripheral Receptor for Cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.-R.; Laviolette, M.; Flamand, N. The CB2 Receptor and Its Role as a Regulator of Inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [Green Version]
- Yang, P.; Myint, K.-Z.; Tong, Q.; Feng, R.; Cao, H.; Almehizia, A.A.; Alqarni, M.H.; Wang, L.; Bartlow, P.; Gao, Y.; et al. Lead Discovery, Chemistry Optimization, and Biological Evaluation Studies of Novel Biamide Derivatives as CB2 Receptor Inverse Agonists and Osteoclast Inhibitors. J. Med. Chem. 2012, 55, 9973–9987. [Google Scholar] [CrossRef] [Green Version]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; Le Fur, G.; Casellas, P. Expression of Central and Peripheral Cannabinoid Receptors in Human Immune Tissues and Leukocyte Subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Idris, A.I.; van ‘t Hof, R.J.; Greig, I.R.; Ridge, S.A.; Baker, D.; Ross, R.A.; Ralston, S.H. Regulation of Bone Mass, Bone Loss and Osteoclast Activity by Cannabinoid Receptors. Nat. Med. 2005, 11, 774–779. [Google Scholar] [CrossRef] [Green Version]
- Contino, M.; Capparelli, E.; Colabufo, N.A.; Bush, A.I. Editorial: The CB2 Cannabinoid System: A New Strategy in Neurodegenerative Disorder and Neuroinflammation. Front. Neurosci. 2017, 11, 196. [Google Scholar] [CrossRef] [Green Version]
- Guindon, J.; Hohmann, A.G. Cannabinoid CB2 Receptors: A Therapeutic Target for the Treatment of Inflammatory and Neuropathic Pain. Br. J. Pharmacol. 2008, 153, 319–334. [Google Scholar] [CrossRef] [Green Version]
- Lunn, C.A.; Reich, E.-P.; Fine, J.S.; Lavey, B.; Kozlowski, J.A.; Hipkin, R.W.; Lundell, D.J.; Bober, L. Biology and Therapeutic Potential of Cannabinoid CB2 Receptor Inverse Agonists. Br. J. Pharmacol. 2008, 153, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol Lipase Regulates Cannabinoid Receptor 2-Dependent Macrophage Activation and Cancer Progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Zhou, S.; Yang, P.; Tian, Y.; Feng, Z.; Xie, X.-Q.; Liu, Y. Targeted Inhibition of the Type 2 Cannabinoid Receptor Is a Novel Approach to Reduce Renal Fibrosis. Kidney Int. 2018, 94, 756–772. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, L.M.; Burford, N.T.; Liao, Y.-H.; Scott, C.E.; Hine, A.M.; Dowling, C.; Chin, J.; Power, M.; Hunnicutt, E.J., Jr.; Emerick, V.L.; et al. Discovery of Selective Cannabinoid CB2 Receptor Agonists by High-Throughput Screening. SLAS Discov. 2018, 23, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Hua, T.; Li, X.; Wu, L.; Iliopoulos-Tsoutsouvas, C.; Wang, Y.; Wu, M.; Shen, L.; Brust, C.A.; Nikas, S.P.; Song, F.; et al. Activation and Signaling Mechanism Revealed by Cannabinoid Receptor-Gi Complex Structures. Cell 2020, 180, 655–665.e18. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Hua, T.; Vemuri, K.; Ho, J.-H.; Wu, Y.; Wu, L.; Popov, P.; Benchama, O.; Zvonok, N.; Locke, K.; et al. Crystal Structure of the Human Cannabinoid Receptor CB2. Cell 2019, 176, 459–467.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazieva, G.A.; Kravchenko, A.N.; Lebedev, O.V.; Strelenko, Y.A.; Chegaev, K.Y. Reactions of Sulfonamides with 4,5-Dihydroxyimidazolidin-2-Ones. Russ. Chem. Bull. 1998, 47, 1561–1564. [Google Scholar] [CrossRef]
- Ohta, H.; Ishizaka, T.; Yoshinaga, M.; Morita, A.; Tomishima, Y.; Toda, Y.; Saito, S. Sulfonamide Derivatives as New Potent and Selective CB2 Cannabinoid Receptor Agonists. Bioorg. Med. Chem. Lett. 2007, 17, 5133–5135. [Google Scholar] [CrossRef]
- Mugnaini, C.; Kostrzewa, M.; Bryk, M.; Mahmoud, A.M.; Brizzi, A.; Lamponi, S.; Giorgi, G.; Ferlenghi, F.; Vacondio, F.; Accioni, P.; et al. Design, Synthesis, and Physicochemical and Pharmacological Profiling of 7-Hydroxy-5-oxopyrazolo[4,3-b]pyridine-6-carboxamide Derivatives with Activity In Vivo. J. Med. Chem. 2020, 63, 7369–7391. [Google Scholar] [CrossRef]
- Rinaldi-Carmona, M.; Barth, F.; Millan, J.; Derocq, J.M.; Casellas, P.; Congy, C.; Oustric, D.; Sarran, M.; Bouaboula, M.; Calandra, B.; et al. SR 144528, the first potent and selective antagonist of the CB2 cannabinoid receptor. J. Pharmacol. Exp. Ther. 1998, 284, 644–650. [Google Scholar]
- Kruse, A.C.; Hu, J.; Pan, A.C.; Arlow, D.H.; Rosenbaum, D.M.; Rosemond, E.; Green, H.F.; Liu, T.; Chae, P.S.; Dror, R.O.; et al. Structure and Dynamics of the M3 Muscarinic Acetylcholine Receptor. Nature 2012, 482, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Kruse, A.C.; Ring, A.M.; Manglik, A.; Hu, J.; Hu, K.; Eitel, K.; Hübner, H.; Pardon, E.; Valant, C.; Sexton, P.M.; et al. Activation and Allosteric Modulation of a Muscarinic Acetylcholine Receptor. Nature 2013, 504, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thal, D.M.; Sun, B.; Feng, D.; Nawaratne, V.; Leach, K.; Felder, C.C.; Bures, M.G.; Evans, D.A.; Weis, W.I.; Bachhawat, P.; et al. Crystal Structures of the M1 and M4 Muscarinic Acetylcholine Receptors. Nature 2016, 531, 335–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.F.; Noinaj, N.; Shibata, Y.; Love, J.; Kloss, B.; Xu, F.; Gvozdenovic-Jeremic, J.; Shah, P.; Shiloach, J.; Tate, C.G.; et al. Structure of the Agonist-Bound Neurotensin Receptor. Nature 2012, 490, 508–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Linciano, P.; Gianquinto, E.; Montanari, M.; Maso, L.; Bellio, P.; Cebrián-Sastre, E.; Celenza, G.; Blázquez, J.; Cendron, L.; Spyrakis, F.; et al. 4-Amino-1,2,4-Triazole-3-Thione as a Promising Scaffold for the Inhibition of Serine and Metallo-β-Lactamases. Pharmaceuticals 2020, 13, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spyrakis, F.; Cellini, B.; Bruno, S.; Benedetti, P.; Carosati, E.; Cruciani, G.; Micheli, F.; Felici, A.; Cozzini, P.; Kellogg, G.E.; et al. Targeting Cystalysin, a Virulence Factor of Treponema Denticola-Supported Periodontitis. ChemMedChem 2014, 9, 1501–1511. [Google Scholar] [CrossRef]
- Santucci, M.; Spyrakis, F.; Cross, S.; Quotadamo, A.; Farina, D.; Tondi, D.; De Luca, F.; Docquier, J.-D.; Prieto, A.I.; Ibacache, C.; et al. Computational and Biological Profile of Boronic Acids for the Detection of Bacterial Serine- and Metallo-β-Lactamases. Sci. Rep. 2017, 7, 17716. [Google Scholar] [CrossRef] [Green Version]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Milletti, F.; Storchi, L.; Sforna, G.; Cross, S.; Cruciani, G. Tautomer Enumeration and Stability Prediction for Virtual Screening on Large Chemical Databases. J. Chem. Inf. Model. 2009, 49, 68–75. [Google Scholar] [CrossRef]
- Tonelli, M.; Cichero, E.; Mahmoud, A.M.; Rabbito, A.; Tasso, B.; Fossa, P.; Ligresti, A. Exploring the effectiveness of novel benzimidazoles as CB2 ligands: Synthesis, biological evaluation, molecular docking studies and ADMET prediction. MedChemCom 2018, 9, 2045–2054. [Google Scholar] [CrossRef] [PubMed]
- Lunn, C.A.; Fine, J.; Rojas-Triana, A.; Jackson, J.V.; Lavey, B.; Kozlowski, J.A.; Hipkin, R.W.; Lundell, D.J.; Bober, L. Cannabinoid CB(2)-selective inverse agonist protects against antigen-induced bone loss. Immunopharmacol. Immunotoxicol. 2007, 29, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Franks, L.N.; Ford, B.M.; Fujiwara, T.; Zhao, H.; Prathera, P.L. The tamoxifen derivative ridaifen-B is a high affinity selective CB2 receptor inverse agonist exhibiting anti-inflammatory and anti-osteoclastogenic effects. Toxicol. Appl. Pharmacol. 2018, 353, 31–42. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
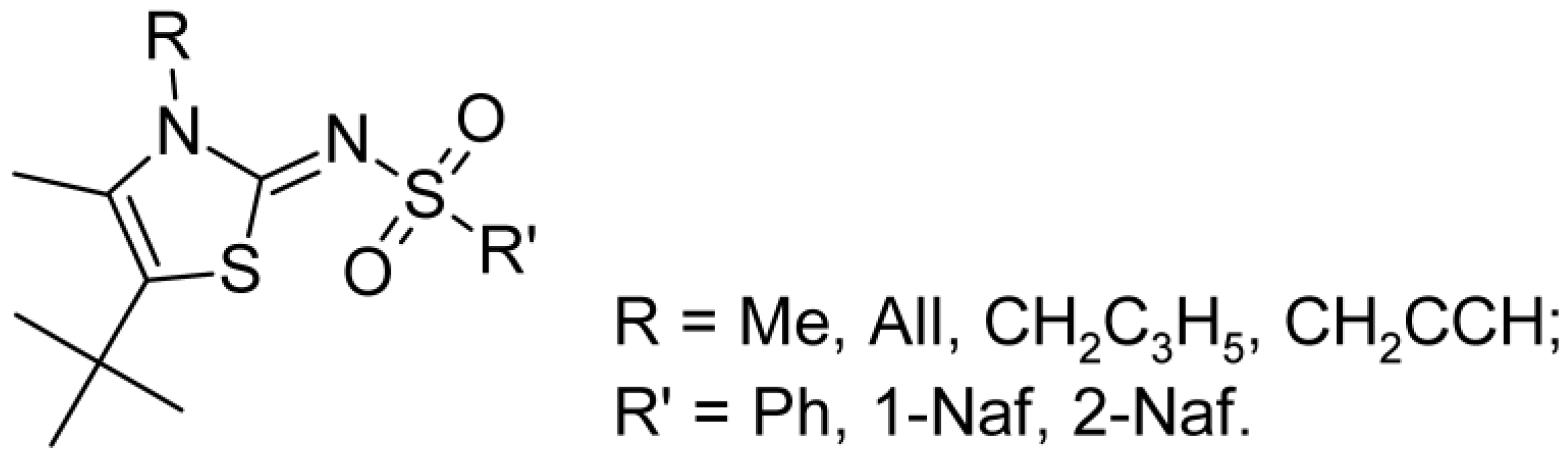{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Compounds | R | R′ | R″ | Ki hCB2R (µM ± S.E.) 1 [% Displacement/ Max Conc. Tested] | Ki hCB1R (µM ± S.E.) 1 [% Displacement/ Max. Conc. Tested] |
| 1 | Et | Et | 4-CH3C6H4 | >10 [7.70 ± 7.58%/ 10µM] | >10 [8.78 ± 3.78%/ 10 µM] |
| 4a | Me | Me | Ph | >10 [6.19 ± 0.98%/ 10 µM] | >10 [32.82 ± 8.18%/ 10 µM] |
| 4b | Me | Me | 4-CH3C6H4 | >10 [7.08 ± 2.03%/ 10 µM] | >10 [26.93 ± 5.68%/ 10µM] |
| 4c | Me | Me | 4-ClC6H4 | >10 [12.35 ± 2.36%/ 10 µM] | >10 [47.52 ± 1.02%/ 10 µM] |
| 4d | Me | Me | 4-BrC6H4 | >10 [10.66 ± 4.33%/ 10 µM] | >10 [14.86 ± 14.85%/ 10 µM] |
| 4e | Et | Et | Ph | >10 [7.72 ± 3.05%/ 10 µM] | >10 [22.68 ± 0.47%/ 10 µM] |
| 4f | Et | Et | 4-ClC6H4 | >10 [14.87 ± 11.90%/ 10 µM] | >10 [17.81 ± 0.06%/ 10 µM] |
| 4g | Et | Et | 4-BrC6H4 | >10 [13.38 ± 8.94%/ 10 µM] | >10 [20.62 ± 1.12%/ 10 µM] |
| 9a | t-Bu | Ph | CH3 | >10 [8.84 ± 1.67%/ 10 µM] | >10 [9.48 ± 5.73%/ 10 µM] |
| 9b | t-Bu | Ph | Ph | >10 [22.61 ± 8.27%/ 10 µM] | >10 [11.97 ± 4.75%/ 10 µM] |
| 9c | t-Bu | Ph | 4-CH3C6H4 | >10 [5.39 ± 1.96%/ 10 µM] | >10 [7.22 ± 1.22%/ 10 µM] |
| 9d | t-Bu | Ph | 4-BrC6H4 | >10 [53.51 ± 8.55%/ 10 µM] | >10 [17.44 ± 0.20%/ 10 µM] |
| 9e | t-Bu | Ph | 3-BrC6H4 | >10 [24.07 ± 12.54%/ 10 µM] | >10 [4.64 ± 4.60%/ 10 µM] |
| 9f | t-Bu | Ph | 2,4,6-(CH3)3C6H2 | >10 [47.77 ± 13.48%/ 10 µM] | >10 [0.00 ± 0.00%/ 10 µM] |
| 9g | t-Bu | Ph | 1-Naf | >10 [11.02 ± 2.06%/10 µM] | >10 [14.85 ± 0.85%/10 µM] |
| 9h | t-Bu | Ph | 2-Naf | >10 [35.64 ± 13.64%/ 10 µM] | >10 [8.35 ± 7.34%/ 10 µM] |
| 10a | t-Bu | CH2(C3H5) | CH3 | >10 [13.36 ± 9.44%/ 10 µM] | >10 [16.31 ± 9.44%/ 10 µM] |
| 10b | t-Bu | CH2(C3H5) | Ph | 1.67 ± 0.16 [73.04 ± 0.80%/ 25 µM] | >10 [10.50 ± 10.30%/ 10 µM] |
| 10c | t-Bu | CH2(C3H5) | 4-CH3C6H4 | 1.47 ± 0.05 [89.48 ± 1.44%/ 25 µM] | >10 [0.00 ± 0.00%/ 10 µM] |
| 10d | t-Bu | CH2(C3H5) | 4-BrC6H4 | 0.83 ± 0.25 [77.23 ± 0.36%/ 10 µM] | >10 [22.25 ± 1.75%/ 10 µM] |
| 10e | t-Bu | CH2(C3H5) | 3-BrC6H4 | 0.34 ± 0.08 [83.53 ± 8.32%/ 10 µM] | >10 [10.78 ± 0.87%/ 10 µM] |
| 10f | t-Bu | CH2(C3H5) | 2,4,6-(CH3)3C6H2 | 0.63 ± 0.14 [81.31 ± 1.31%/ 10 µM] | >10 [15.41 ± 7.13%/ 10 µM] |
| 10g | t-Bu | CH2(C3H5) | 1-Naf | 0.06 ± 0.01 [95.65 ± 2.65%/ 10 µM] | >10 [24.85 ± 2.39% /10 µM] |
| 10h | t-Bu | CH2(C3H5) | 2-Naf | 0.78 ± 0.17 [72.34 ± 5.08%/ 10 µM] | >10 [8.18 ± 3.25%/ 10 µM] |
| 11a | t-Bu | CH2CH2OCH3 | CH3 | >10 [14.09 ± 7.94%/ 10 µM] | >10 [65.33 ± 16.41%/ 10 µM] |
| 11b | t-Bu | CH2CH2OCH3 | Ph | >10 [21.22 ± 13.17%/ 10 µM] | >10 [52.56 ± 5.88%/ 10 µM] |
| 11c | t-Bu | CH2CH2OCH3 | 4-CH3C6H4 | >10 [33.36 ± 7.52%/ 10 µM] | >10 [57.05 ± 32.82%/ 10 µM] |
| 11d | t-Bu | CH2CH2OCH3 | 4-BrC6H4 | >10 [42.69 ± 8.61%/ 10 µM] | >10 [50.15 ± 15.45%/ 10 µM] |
| 11e | t-Bu | CH2CH2OCH3 | 3-BrC6H4 | >10 [41.09 ± 5.54%/ 10 µM] | >10 [10.00 ± 10.00%/ 10 µM] |
| 11f | t-Bu | CH2CH2OCH3 | 2,4,6-(CH3)3C6H2 | 3.45 ± 0.95 [64.83 ± 0.41%/ 10 µM] | >10 [55.66 ± 32.59%/ 10 µM] |
| 11g | t-Bu | CH2CH2OCH3 | 1-Naf | 1.66 ± 0.20 [78.73 ± 6.26%/ 10 µM] | >10 [13.50 ± 3.51%/ 10 µM] |
| 11h | t-Bu | CH2CH2OCH3 | 2-Naf | >10 [40.14 ± 2.48%/ 10 µM] | >10 [45.33 ± 38.80%/ 10 µM] |
| 12a | 1-Ad | CH2(C3H5) | CH3 | 0.28 ± 0.05 [76.50 ± 3.50%/ 25 µM] | 0.210 ± 0.004 [76.52 ± 3.48%/ 25 µM] |
| 12b | 1-Ad | CH2(C3H5) | Ph | 0.26 ± 0.03 [84.50 ± 4.50%/ 25 µM] | >10 [29.47 ± 19.44%/ 25 µM] |
| 12c | 1-Ad | CH2(C3H5) | 4-CH3C6H4 | >10 [20.56 ± 10.56%/ 10 µM] | >10 [34.64 ± 8.01%/ 10 µM] |
| 12d | 1-Ad | CH2(C3H5) | 4-BrC6H4 | >10 [32.52 ± 8.15%/ 10 µM] | >10 [47.85 ± 16.24%/ 10 µM] |
| 12e | 1-Ad | CH2(C3H5) | 3-BrC6H4 | 0.16 ± 0.03 [71.39 ± 0.60%/ 10 µM] | >10 [5.07 ± 3.40%/ 10 µM] |
| 12f | 1-Ad | CH2(C3H5) | 2,4,6-(CH3)3C6H2 | >10 [40.25 ± 6.92%/ 10 µM] | >10 [59.18 ± 5.97%/ 10 µM] |
| 12g | 1-Ad | CH2(C3H5) | 1-Naf | >10 [45.58 ± 3.43%/ 10 µM] | >10 [12.79 ± 0.87%/ 10 µM] |
| 12h | 1-Ad | CH2(C3H5) | 2-Naf | >10 [17.72 ± 11.38%/ 10 µM] | >10 [38.54 ± 13.35%/ 10 µM] |
| 13 | i-Pr | CH2(C3H5) | 1-Naf | 0.80 ± 0.02 [78.27 ± 1.73%/ 10 µM] | >10 [4.63 ± 4.59%/ 10 µM] |
| 14 | t-Bu | i-Bu | 4-CH3C6H4 | >10 [47.77 ± 13.48%/ 10 µM] | >10 [0.02 ± 0.01%/ 10 µM] |
| 15 | CH2(C3H5) | t-Bu | Ph | 1.61 ± 0.16 [73.92 ± 0.92%/ 10 µM] | >10 [0.01 ± 0.01%/ 10 µM] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianquinto, E.; Sodano, F.; Rolando, B.; Kostrzewa, M.; Allarà, M.; Mahmoud, A.M.; Kumar, P.; Spyrakis, F.; Ligresti, A.; Chegaev, K. N-[1,3-Dialkyl(aryl)-2-oxoimidazolidin-4-ylidene]-aryl(alkyl)sulphonamides as Novel Selective Human Cannabinoid Type 2 Receptor (hCB2R) Ligands; Insights into the Mechanism of Receptor Activation/Deactivation. Molecules 2022, 27, 8152. https://doi.org/10.3390/molecules27238152
Gianquinto E, Sodano F, Rolando B, Kostrzewa M, Allarà M, Mahmoud AM, Kumar P, Spyrakis F, Ligresti A, Chegaev K. N-[1,3-Dialkyl(aryl)-2-oxoimidazolidin-4-ylidene]-aryl(alkyl)sulphonamides as Novel Selective Human Cannabinoid Type 2 Receptor (hCB2R) Ligands; Insights into the Mechanism of Receptor Activation/Deactivation. Molecules. 2022; 27(23):8152. https://doi.org/10.3390/molecules27238152
Chicago/Turabian StyleGianquinto, Eleonora, Federica Sodano, Barbara Rolando, Magdalena Kostrzewa, Marco Allarà, Ali Mokhtar Mahmoud, Poulami Kumar, Francesca Spyrakis, Alessia Ligresti, and Konstantin Chegaev. 2022. "N-[1,3-Dialkyl(aryl)-2-oxoimidazolidin-4-ylidene]-aryl(alkyl)sulphonamides as Novel Selective Human Cannabinoid Type 2 Receptor (hCB2R) Ligands; Insights into the Mechanism of Receptor Activation/Deactivation" Molecules 27, no. 23: 8152. https://doi.org/10.3390/molecules27238152