
Stabilizing Halogen-Bonded Complex between Metallic Anion and Iodide

Abstract
:1. Introduction
2. Methods
2.1. Experimental Methods
2.2. Computational Methods
3. Results and Discussion
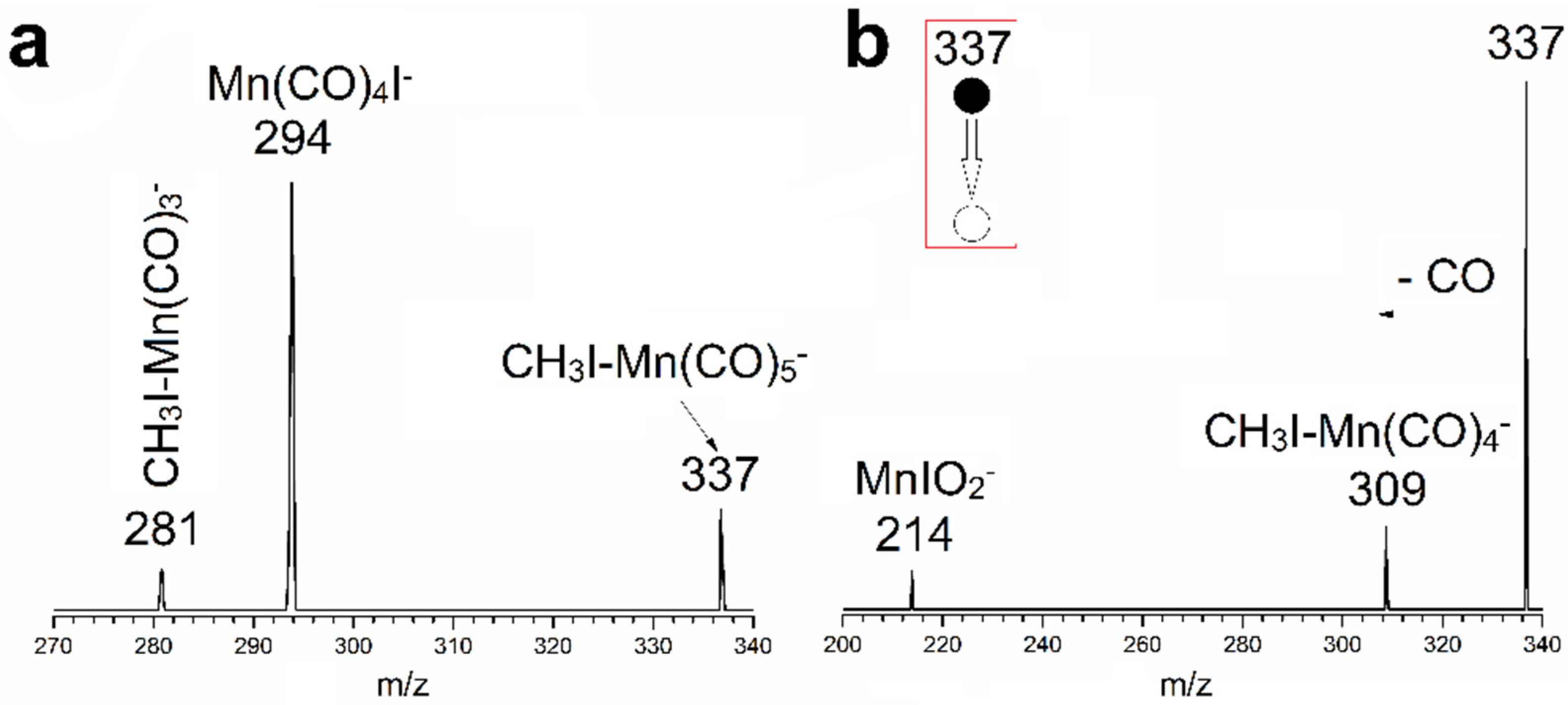
3.1. Mass Spectrometry
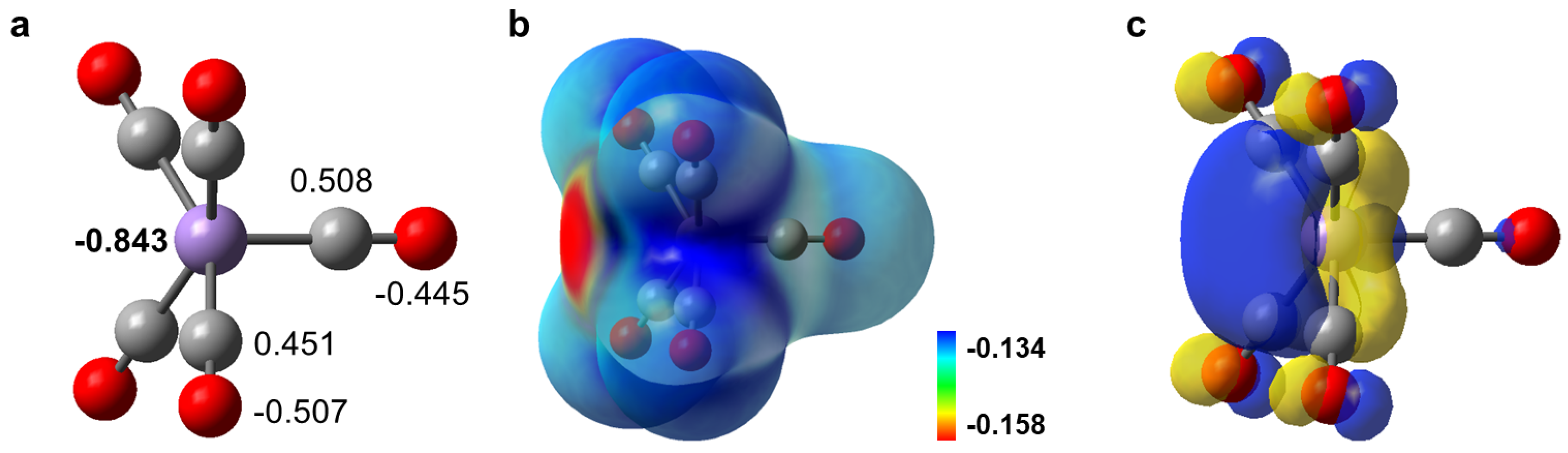
3.2. Density Functional Theory Calculation
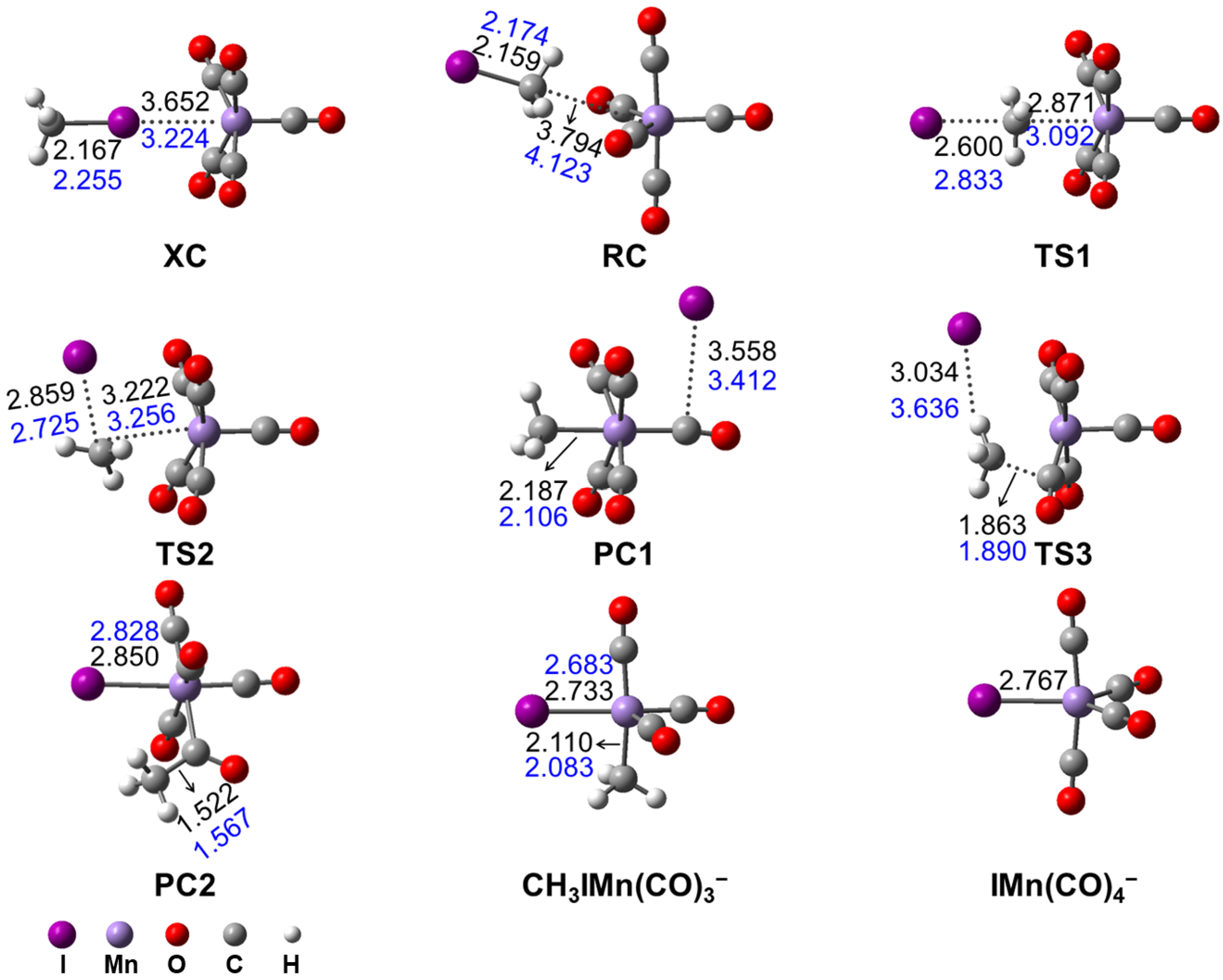
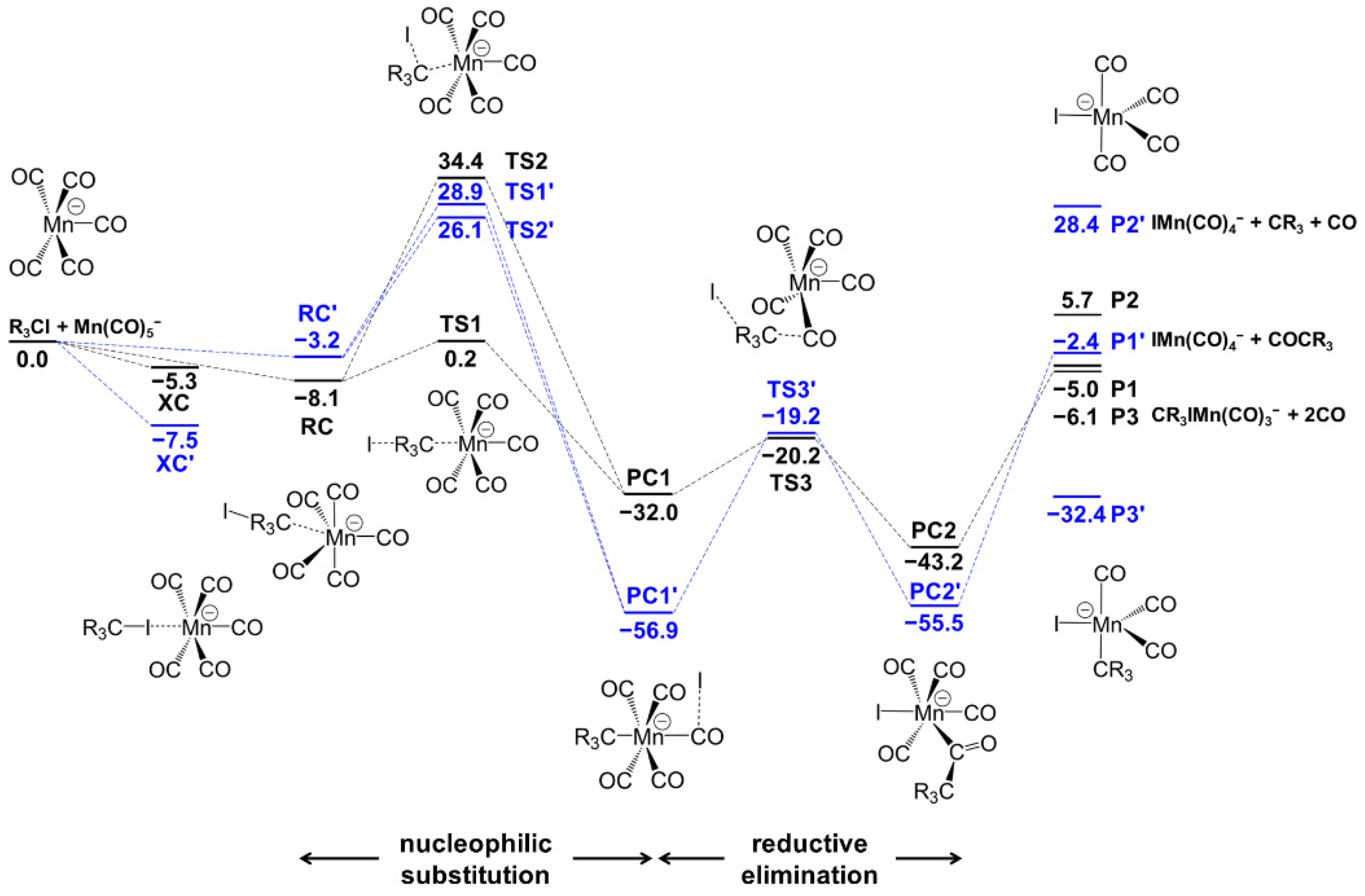
3.2.1. Mn(CO)5− + CH3I Reaction Mechanism
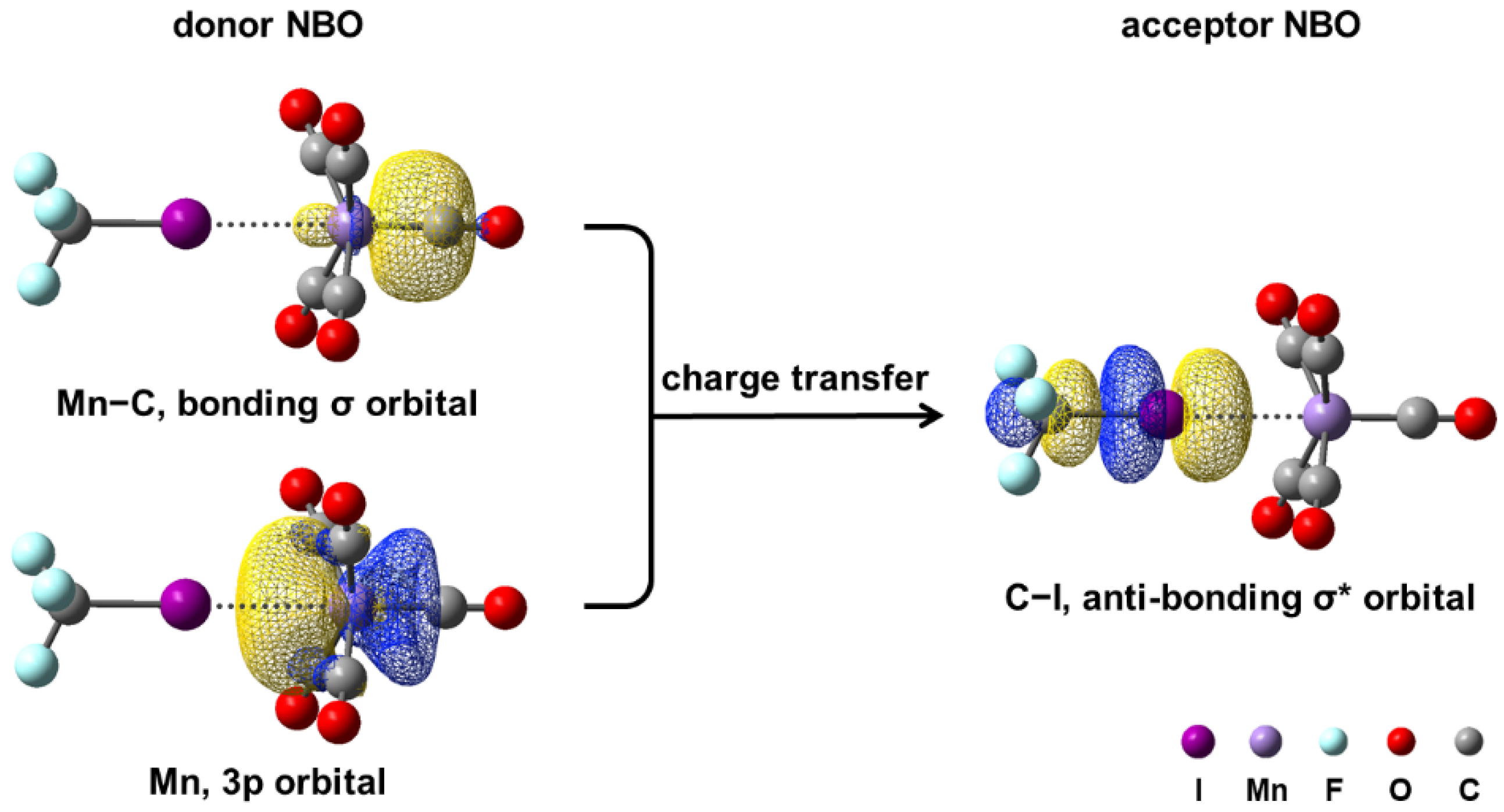
3.2.2. Stabilizing Halogen-Bonded Complex by CF3I
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gou, Q.; Feng, G.; Evangelisti, L.; Vallejo-López, M.; Spada, L.; Lesarri, A.; Cocinero, E.J.; Caminati, W. Internal Dynamics in Halogen-Bonded Adducts: A Rotational Study of Chlorotrifluoromethane–Formaldehyde. Chem. Eur. J. 2015, 21, 4148–4152. [Google Scholar] [CrossRef] [PubMed]
- Inscoe, B.; Rathnayake, H.; Mo, Y. Role of Charge Transfer in Halogen Bonding. J. Phys. Chem. A 2021, 125, 2944–2953. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Zheng, Y.; Feng, G.; Gou, Q. Halogen Bond in the Water Adduct of Chloropentafluoroethane Revealed by Rotational Spectroscopy. J. Chem. Phys. 2018, 149, 154307. [Google Scholar] [CrossRef] [PubMed]
- Kolář, M.H.; Hobza, P. Computer Modeling of Halogen Bonds and Other σ-Hole Interactions. Chem. Rev. 2016, 116, 5155–5187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, C.C.; Wright, J.S.; Carrington, E.J.; Perutz, R.N.; Hunter, C.A.; Brammer, L. Hydrogen Bonding vs. Halogen Bonding: The Solvent Decides. Chem. Sci. 2017, 8, 5392–5398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabó, I.; Olasz, B.; Czakó, G. Deciphering Front-Side Complex Formation in SN2 Reactions via Dynamics Mapping. J. Phys. Chem. Lett. 2017, 8, 2917–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Herbers, S.; Gou, Q.; Caminati, W.; Grabow, J.U. Chlorine “Equatorial Belt” Activation of CF3Cl by CO2: The C···Cl Tetrel Bond Dominance in CF3Cl–CO2. J. Phys. Chem. Lett. 2021, 12, 3907–3913. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Ho, P.S.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the Halogen Bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Clark, T. σ-Holes. WIREs Comput. Mol. Sci. 2013, 3, 13–20. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen Bonding: The σ-Hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Auffinger, P.; Hays, F.A.; Westhof, E.; Ho, P.S. Halogen Bonds in Biological Molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 16789–16794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilday, L.C.; Robinson, S.W.; Barendt, T.A.; Langton, M.J.; Mullaney, B.R.; Beer, P.D. Halogen Bonding in Supramolecular Chemistry. Chem. Rev. 2015, 115, 7118–7195. [Google Scholar] [CrossRef]
- Mukherjee, A.; Tothadi, S.; Desiraju, G.R. Halogen Bonds in Crystal Engineering: Like Hydrogen Bonds yet Different. Acc. Chem. Res. 2014, 47, 2514–2524. [Google Scholar] [CrossRef]
- Priimagi, A.; Cavallo, G.; Metrangolo, P.; Resnati, G. The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances. Acc. Chem. Res. 2013, 46, 2686–2695. [Google Scholar] [CrossRef] [Green Version]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen Interactions in Protein–Ligand Complexes: Implications of Halogen Bonding for Rational Drug Design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, G.; Ciborowski, S.; Bowen, K. Stabilizing Otherwise Unstable Anions with Halogen Bonding. Angew. Chem. Int. Ed. 2017, 56, 9897–9900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muramatsu, S.; Koyasu, K.; Tsukuda, T. Formation of Grignard Reagent-like Complex CH3−M−I− via Oxidative Addition of CH3I on Coinage Metal Anions M− (M = Cu, Ag, Au) in the Gas Phase. Chem. Lett. 2017, 46, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, S.; Koyasu, K.; Tsukuda, T. Oxidative Addition of CH3I to Au– in the Gas Phase. J. Phys. Chem. A 2016, 120, 957–963. [Google Scholar] [CrossRef]
- Wang, F.; Ji, X.; Ying, F.; Zhang, J.; Zhao, C.; Xie, J. Computational Studies of Coinage Metal Anion M− + CH3X (X = F, Cl, Br, I) Reactions in Gas Phase. Molecules 2022, 27, 307. [Google Scholar] [CrossRef]
- Zhang, X.; Bowen, K. Designer Metallic Acceptor-Containing Halogen Bonds: General Strategies. Chem. Eur. J. 2017, 23, 5439–5442. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Dunning, T.H., Jr. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Balabanov, N.B.; Peterson, K.A. Systematically Convergent Basis Sets for Transition Metals. I. All-Electron Correlation Consistent Basis Sets for the 3d Elements Sc–Zn. J. Chem. Phys. 2005, 123, 064107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulliken, R.S. Electronic Population Analysis on LCAO–MO Molecular Wave Functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically Convergent Basis Sets with Relativistic Pseudopotentials. II. Small-Core Pseudopotentials and Correlation Consistent Basis Sets for the Post-d Group 16–18 Elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A.; Shepler, B.C.; Figgen, D.; Stoll, H. On the Spectroscopic and Thermochemical Properties of ClO, BrO, IO, and Their Anions. J. Phys. Chem. A 2006, 110, 13877–13883. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mensa-Bonsu, G.; Tozer, D.J.; Verlet, J.R.R. Photoelectron Spectroscopic Study of I−·ICF3: A Frontside Attack SN2 Pre-Reaction Complex. Phys. Chem. Chem. Phys. 2019, 21, 13977–13985. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular Interactions from a Natural Bond Orbital, Donor-Acceptor Viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural Population Analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Ji, X.; Zhao, C.; Xie, J. Investigating the Role of Halogen-Bonded Complexes in Microsolvated Y−(H2O)n + CH3I SN2 reactions. Phys. Chem. Chem. Phys. 2021, 23, 6349–6360. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| kcal/mol | ∆Eelec | ∆(Eelec + ZPE) | ∆H298.15K | ∆G298.15K | ||||
|---|---|---|---|---|---|---|---|---|
| R | H | F | H | F | H | F | H | F |
| Mn(CO)5− + CR3I | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| XC | −5.4 | −17.5 | −5.3 | −17.5 | −3.9 | −16.5 | 1.4 | −7.0 |
| RC | −8.6 | −3.6 | −9.1 | −3.2 | −7.3 | −1.8 | −0.2 | 3.8 |
| TS1 | 0.4 | 29.1 | 0.2 | 28.9 | 1.2 | 30.3 | 8.6 | 37.3 |
| TS2 | 35.2 | 27.2 | 34.4 | 26.1 | 35.4 | 27.6 | 43.0 | 35.2 |
| PC1 | −31.7 | −56.6 | −32.0 | −56.9 | −31.1 | −55.1 | −22.4 | −47.6 |
| TS3 | −20.3 | −18.8 | −20.2 | −19.2 | −19.5 | −17.8 | −10.4 | −9.9 |
| PC3 | −44.4 | −56.7 | −43.2 | −55.5 | −42.4 | −54.9 | −33.3 | −43.5 |
| Mn(CO)4I− + COCR3 | −3.0 | −0.7 | −5.0 | −2.4 | −3.9 | −1.4 | −8.2 | −5.8 |
| Mn(CO)4I− + CO + CR3 | 13.1 | 33.4 | 5.7 | 28.4 | 8.4 | 30.2 | −4.7 | 16.3 |
| CR3I−Mn(CO)3− + 2CO | −1.6 | −28.3 | −6.1 | −32.4 | −4.4 | −29.7 | −15.5 | −43.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ying, F.; Yuan, X.; Zhang, X.; Xie, J. Stabilizing Halogen-Bonded Complex between Metallic Anion and Iodide. Molecules 2022, 27, 8069. https://doi.org/10.3390/molecules27228069
Ying F, Yuan X, Zhang X, Xie J. Stabilizing Halogen-Bonded Complex between Metallic Anion and Iodide. Molecules. 2022; 27(22):8069. https://doi.org/10.3390/molecules27228069
Chicago/Turabian StyleYing, Fei, Xu Yuan, Xinxing Zhang, and Jing Xie. 2022. "Stabilizing Halogen-Bonded Complex between Metallic Anion and Iodide" Molecules 27, no. 22: 8069. https://doi.org/10.3390/molecules27228069