
SNH Amidation of 5-Nitroisoquinoline: Access to Nitro- and Nitroso Derivatives of Amides and Ureas on the Basis of Isoquinoline

Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Bala, M.; Kumar, S.; Pratap, K.; Verma, P.K.; Padwad, Y.; Singh, B. Bioactive isoquinoline alkaloids from Cissampelos pareira. Nat. Prod. Res. 2019, 33, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.-Q.; Bi, J.-L.; Sun, Q.-Y.; Yang, F.-M.; Wang, Y.-H.; Tang, G.-H.; Zhao, F.-W.; Wang, H.; Xu, J.-J.; Kennelly, E.J.; et al. Bioactive isoquinoline alkaloids from Corydalis saxicola. Planta Med. 2012, 78, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, P.; Kumar, G.S. Isoquinoline alkaloids and their binding with polyadenylic acid: Potential basis of therapeutic action. Mini Rev. Med. Chem. 2010, 10, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Wireko, M.A.; Wang, H.; Wu, C.; Wang, Q.; Zhang, H.; Cao, Y. Isoquinolines: Important Cores in Many Marketed and Clinical Drugs. Anti-Cancer Agents Med. Chem. 2021, 21, 811–824. [Google Scholar] [CrossRef]
- Jaryaram, V.; Sridhar, T.; Sharma, G.V.M.; Berrée, F.; Carboni, B. Synthesis of polysubstituted isoquinolines and related fused pyridines from alkenyl boronic esters via a copper-catalyzed azidation/aza-Wittig Condensation Sequence. J. Org. Chem. 2018, 83, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhang, Y.-X.; Nie, X.-D.; Si, C.-M.; Sun, X.; Wei, G.-G. Approach to Chiral 1-Substituted Isoquinolone and 3-Substituted Isoindolin-1-one by Addition–Cyclization Process. J. Org. Chem. 2018, 83, 9879–9889. [Google Scholar] [CrossRef]
- Zhu, Z.; Tang, X.; Li, X.; Wu, W.; Deng, G.; Jiang, H. Palladium-Catalyzed C–H Functionalization of Aromatic Oximes: A Strategy for the Synthesis of Isoquinolines. J. Org. Chem. 2016, 81, 1401–1409. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, M.; Wang, L.; Chen, K.; Wang, J.; Song, C.; Zhu, J. Bidentate directing-enabled, traceless heterocycle synthesis: Cobalt-catalyzed access to isoquinolines. Org. Lett. 2016, 18, 5632–5635. [Google Scholar] [CrossRef]
- Chu, H.; Xue, P.; Yu, J.-T.; Cheng, J. Rhodium-Catalyzed Annulation of Primary Benzylamine with α-Diazo Ketone toward Isoquinoline. J. Org. Chem. 2016, 81, 8009–8013. [Google Scholar] [CrossRef]
- Jacob, J.; Varghese, N.; Rasheed, S.P.; Agnihotri, S.; Sharma, V.; Wakode, S. Recent advances in the synthesis of isoquinoline and its analogue: A review. WJPPS 2016, 5, 1821–1837. [Google Scholar] [CrossRef]
- Yang, D.; Burugupalli, S.; Daniel, D.; Chen, Y. Microwave-assisted one-pot synthesis of isoquinolines, furopyridines, and thienopyridines by palladium-catalyzed sequential Coupling–Imination–Annulation of 2-bromoarylaldehydes with terminal acetylenes and ammonium acetate. J. Org. Chem. 2012, 77, 4466–4472. [Google Scholar] [CrossRef]
- Zheng, L.; Ju, J.; Bin, Y.; Hua, R. Synthesis of isoquinolines and heterocycle-fused pyridines via three-component cascade reaction of aryl ketones, hydroxylamine, and alkynes. J. Org. Chem. 2012, 77, 5794–5800. [Google Scholar] [CrossRef]
- Chinnagolla, R.K.; Pimparkar, S.; Jeganmohan, M. Ruthenium-Catalyzed Highly Regioselective Cyclization of Ketoximes with Alkynes by C–H Bond Activation: A Practical Route to Synthesize Substituted Isoquinolines. Org. Lett. 2012, 14, 3032–3035. [Google Scholar] [CrossRef]
- Clarke, P.A.; Santos, S.; Martin, W.H.C. Combining pot, atom and step economy (PASE) in organic synthesis. Synthesis of tetrahydropyran-4-ones. Green Chem. 2007, 9, 438–440. [Google Scholar] [CrossRef] [Green Version]
- Arends, I.; Sheldon, R.; Hanefeld, U. Green Chemistry and Catalysis; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Charushin, V.N.; Chupakhin, O.N. Nucleophilic C—H functionalization of arenes: A contribution to green chemistry. Russ. Chem. Bull. 2019, 68, 453–471. [Google Scholar] [CrossRef]
- Chupakhin, O.N.; Charushin, V.N. Recent advances in the field of nucleophilic aromatic substitution of hydrogen. Tetrahedron Lett. 2016, 57, 2665–2672. [Google Scholar] [CrossRef]
- Charushin, V.N.; Chupakhin, O.N. Metal-free C–H functionalization of aromatic compounds through the action of nucleophilic reagents. Top. Heterocycl. Chem. 2014, 37, 1–50. [Google Scholar] [CrossRef]
- Gulevskaya, A.V.; Pozharskii, A.F. The SNH-Amination of Heteroaromatic Compounds. Top. Heterocycl. Chem. 2013, 37, 179–239. [Google Scholar] [CrossRef]
- Budyka, M.F.; Terent’ev, P.B.; Kost, A.N. Amination of 5-azacinnoline with aromatic amines. Chem. Heterocycl. Compd. 1978, 14, 663–666. [Google Scholar] [CrossRef]
- Borovlev, I.V.; Demidov, O.P.; Saigakova, N.A.; Amangasieva, G.A. SNH-and SNipso-Arylamination of 1, 3, 7-Triazapyrenes. Eur. J. Org. Chem. 2014, 2014, 7675–7683. [Google Scholar] [CrossRef]
- Shchepochkin, A.V.; Chupakhin, O.N.; Charushin, V.N.; Steglenko, D.V.; Minkin, V.I.; Rusinov, G.L.; Matern, A.I. C–H functionalization of azines. Anodic dehydroaromatization of 9-(hetero) aryl-9, 10-dihydroacridines. RSC Adv. 2016, 6, 77834–77840. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Lushchekina, S.V.; Boltneva, N.P.; Serebryakova, O.G.; Rudakova, E.V.; Ustyugov, A.A.; Bachurin, S.O.; Shchepochkin, A.V.; Chupakhin, O.N.; Charushin, V.N.; et al. 9-Substituted acridine derivatives as acetylcholinesterase and butyrylcholinesterase inhibitors possessing antioxidant activity for Alzheimer’s disease treatment. Bioorg. Med. Chem. 2017, 25, 5981–5994. [Google Scholar] [CrossRef] [PubMed]
- Shchepochkin, A.V.; Chupakhin, O.N.; Charushin, V.N.; Rusinov, G.L.; Subbotina, Y.O.; Slepukhin, P.A.; Budnikova, Y.G. Stable σH-adducts in the reactions of the acridinium cation with heterocyclic N-nucleophiles. Russ. Chem. Bull. 2013, 62, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Gulevskaya, A.V.; Tyaglivaya, I.N.; Verbeeck, S.; Maes, B.U.W.; Tkachuk, A.V. Oxidative arylamination of 1, 3-dinitrobenzene and 3-nitropyridine under anaerobic conditions: The dual role of the nitroarenes. Arkivoc 2011, 2011, 238–251. [Google Scholar] [CrossRef] [Green Version]
- Garnier, E.; Audoux, J.; Pasquinet, E.; Suzenet, F.; Poullain, D.; Lebret, B.; Guillaumet, G. Easy access to 3-or 5-heteroarylamino-1, 2, 4-triazines by SNAr, SNH, and palladium-catalyzed N-heteroarylations. J. Org. Chem. 2004, 69, 7809–7815. [Google Scholar] [CrossRef]
- Matern, A.I.; Charushin, V.N.; Chupakhin, O.N. Progress in the studies of oxidation of dihydropyridines and their analogues. Russ. Chem. Rev. 2007, 76, 23–40. [Google Scholar] [CrossRef]
- Makosza, M.; Wojciechowski, K. Nucleophilic Substitution of Hydrogen in Arenes and Heteroarenes. Top. Heterocycl. Chem. 2013, 37, 51–105. [Google Scholar] [CrossRef]
- Makosza, M. How does nucleophilic aromatic substitution in nitroarenes really proceed: General mechanism. Synthesis 2017, 49, 3247–3254. [Google Scholar] [CrossRef]
- Wróbel, Z.; Kwast, A. 2-Nitroso-N-arylanilines: Products of acid-promoted transformation of σH adducts of arylamines and nitroarenes. Synlett 2007, 10, 1525–1528. [Google Scholar] [CrossRef]
- Wróbel, Z.; Kwast, A. Simple synthesis of N-aryl-2-nitrosoanilines in the reaction of nitroarenes with aniline anion derivatives. Synthesis 2010, 2010, 3865–3872. [Google Scholar] [CrossRef]
- Kwast, A.; Stachowska, K.; Trawczyński, A.; Wróbel, Z. N-Aryl-2-nitrosoanilines as intermediates in the synthesis of substituted phenazines from nitroarenes. Tetrahedron Lett. 2011, 52, 6484–6488. [Google Scholar] [CrossRef]
- Wróbel, Z.; Stachowska, K.; Grudzień, K.; Kwast, A. N-Aryl-2-nitrosoanilines as Intermediates in the Two-Step Synthesis of Substituted 1, 2-Diarylbenzimidazoles from Simple Nitroarenes. Synlett 2011, 10, 1439–1443. [Google Scholar] [CrossRef]
- Wróbel, Z.; Wiecław, M.; Bujok, R.; Wojciechowski, K. Synthesis of pyrrolo [3, 2-a] phenazines from 5-nitroindoles and anilines. Monatsh. Chem. 2013, 144, 1847–1853. [Google Scholar] [CrossRef] [Green Version]
- Bashkin, J.K.; Rains, R.; Stern, M. Taking green chemistry from the laboratory to chemical plant. Green Chem. 1999, 1, G41–G43. [Google Scholar] [CrossRef]
- Triplett, R.D.; Rains, R.K. Process for Preparing 4-aminodiphenylamine Intermediates. US Patent 7504539 B2, 17 March 2009. [Google Scholar]
- Patriciu, O.-I.; Finaru, A.-L.; Sandulescu, I.; Guillaumet, G. Synthesis of Nitro N, N′-Dipyridinylamines via Oxidative Nucleophilic Substitution of Hydrogen. Synthesis 2007, 2007, 3868–3876. [Google Scholar] [CrossRef]
- Wozniak, M.; Baranski, A.; Nowak, K.; Poradowska, H. Regioselectivity of the amination of some nitroisoquinolines by liquid ammonia/potassium permanganate. Liebigs Ann. Chem. 1990, 1990, 653–657. [Google Scholar] [CrossRef]
- Woźniak, M.; Nowak, K. Amination of some nitroisoquinolines with liquid methylamine/potassium permanganate. Liebigs Ann. Chem. 1994, 1994, 355–360. [Google Scholar] [CrossRef]
- Demidov, O.P.; Pobedinskaya, D.Y.; Avakyan, E.K.; Amangasieva, G.A.; Borovlev, I.V. SNH Arylamination of Nitroquinolines: Access to Nitro and Nitroso Derivatives of Arylaminoquinolines. Chem. Heterocycl. Compd. 2018, 54, 875–886. [Google Scholar] [CrossRef]
- Stern, M.K.; Cheng, B.K. Amination of nitrobenzene via nucleophilic aromatic substitution for hydrogen: Direct formation of aromatic amide bonds. J. Org. Chem. 1993, 58, 6883–6888. [Google Scholar] [CrossRef]
- Stern, M.K.; Bashkin, K.J. Method of preparing 4-aminodiphenylamine. US Patent 5 117 063, 26 May 1992. [Google Scholar]
- Borovlev, I.V.; Demidov, O.P.; Kurnosova, N.A.; Amangasieva, G.A.; Avakyan, E.K. Direct oxidative SNH amidation of 1,3,7-triazapyrene. Chem. Heterocycl. Compd. 2015, 51, 170–175. [Google Scholar] [CrossRef]
- Demidov, O.P.; Borovlev, I.V.; Amangasieva, G.A.; Avakyan, E.K. Oxidative SNH amidation of acridine and tautomerism of N-(acridin-9-yl) benzamides. Chem. Heterocycl. Compd. 2016, 52, 104–109. [Google Scholar] [CrossRef]
- Amangasieva, G.A.; Borovlev, I.V.; Demidov, O.P.; Avakyan, E.K.; Borovleva, A.A. Synthesis of Amides by Nucleophilic Substitution of Hydrogen in 3-Nitropyridine. Russ. J. Org. Chem. 2018, 54, 867–872. [Google Scholar] [CrossRef]
- Amangasieva, G.A.; Avakyan, E.K.; Demidov, O.P.; Borovleva, A.A.; Pobedinskaya, D.Y.; Borovlev, I.V. SNH Amidation of nitroquinolines: Synthesis of amides on the basis of nitro-and nitrosoquinolines. Chem. Heterocycl. Compd. 2019, 55, 623–631. [Google Scholar] [CrossRef]
- Amangasieva, G.A.; Borovlev, I.V.; Demidov, O.P.; Kurnosova, N.A.; Avakyan, E.K. Urea in an aminodemethoxylation reaction of 6-methoxy-1,3,7-triazapyrenes. Chem. Heterocycl. Compd. 2015, 51, 586–588. [Google Scholar] [CrossRef]
- Borovlev, I.V.; Demidov, O.P.; Amangasieva, G.A.; Avakyan, E.K.; Kurnosova, N.A. Ureas as new nucleophilic reagents for SNH amination and carbamoyl amination reactions in the 1,3,7-triazapyrene series. ARKIVOC 2016, 2016, 58–70. [Google Scholar] [CrossRef] [Green Version]
- Borovlev, I.V.; Demidov, O.P.; Amangasieva, G.A.; Avakyan, E.K.; Kurnosova, N.A. Ureas as a New Nucleophilic Reagents for SNAr Amination and Carbamoyl Amination Reactions in 1,3,7-Triazapyrene Series. J. Heterocycl. Chem. 2017, 54, 406–412. [Google Scholar] [CrossRef]
- Borovlev, I.V.; Demidov, O.P.; Amangasieva, G.A.; Avakyan, E.K. Direct and facile synthesis of 9-aminoacridine and acridin-9-yl-ureas. Tetrahedron Lett. 2016, 57, 3608–3611. [Google Scholar] [CrossRef]
- Avakyan, E.K.; Borovlev, I.V.; Demidov, O.P.; Amangasieva, G.A.; Pobedinskaya, D.Y. SNH Alkyl carbamoyl amination of 3-nitropyridine: Competitive synthesis of nitro-and nitrosopyridine derivatives. Chem. Heterocycl. Compd. 2017, 53, 1207–1213. [Google Scholar] [CrossRef]
- Demidov, O.P.; Amangasieva, G.A.; Avakyan, E.K.; Borovlev, I.V. Nucleophilic Addition of Amides to 10-Alkylacridinium Cations: A Case of Double N-Nucleophilicity of Some Monoamides. Synthesis 2017, 49, 3710–3719. [Google Scholar] [CrossRef] [Green Version]
- Iriepa, I.; Bellanato, J. Synthesis, spectroscopic, structural and conformational study of some tri-substituted ureas derived from N-methylpiperazine containing phenyl and N-heterocyclic substituents. J. Mol. Struct. 2013, 1044, 215–220. [Google Scholar] [CrossRef]
- Annese, M.; Corradi, A.B.; Forlani, L.; Rizzoli, C.; Sgarabotto, P. Tautomerism in some acetamido derivatives of nitrogen-containing heterocycles: X-ray structural analysis of 2-amino and 2-imino forms of benzothiazole derivatives. J. Chem. Soc. Perkin Trans. 2 1994, 615–621. [Google Scholar] [CrossRef]
- Forlani, L.; Mezzina, E.; Boga, C.; Forconi, M. Tautomerism and Dimerization of Acetamidothiazole Derivatives− UV/Vis and NMR Spectroscopic Investigation. Eur. J. Org. Chem. 2001, 2001, 2779–2785. [Google Scholar] [CrossRef]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Sharp, J.T.; Gosney, I.; Rowley, A.G. Practical Organic Chemistry; Springer: London, UK; New York, NY, USA, 1989; p. 57. [Google Scholar]








{kind=link}
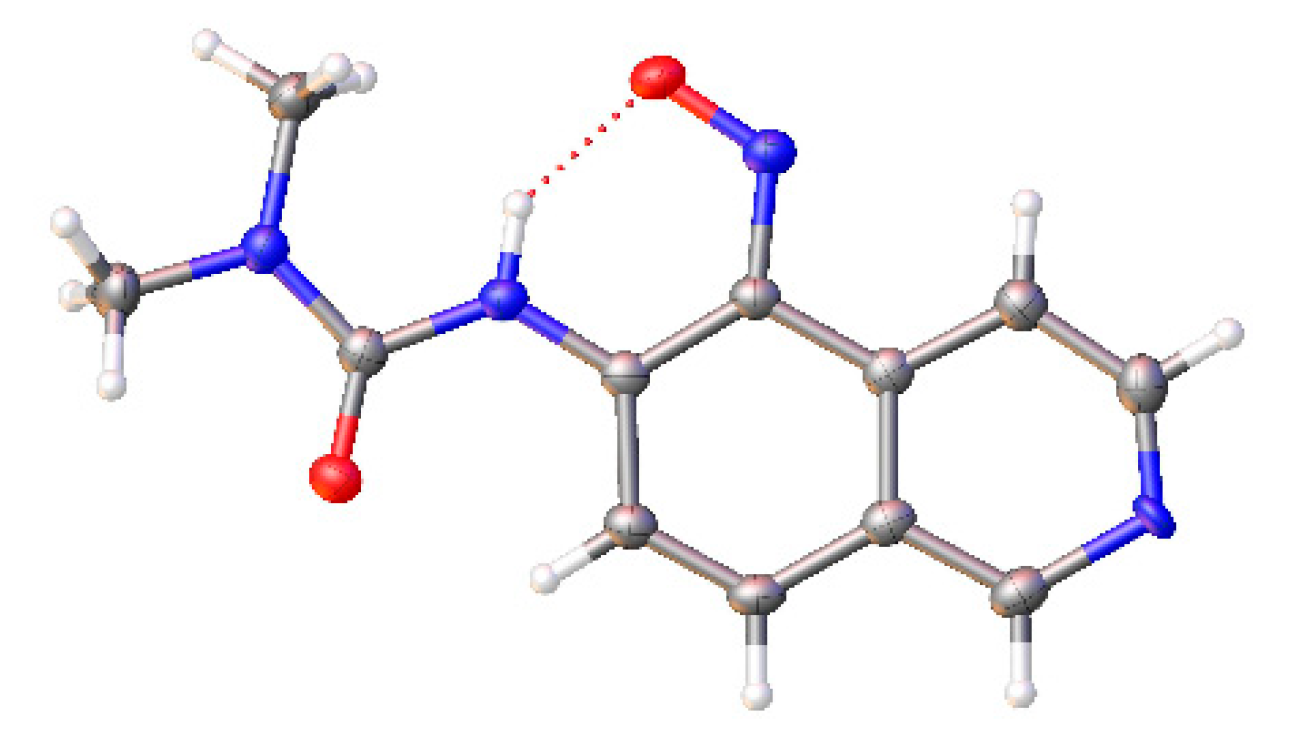{kind=link}
{kind=link}
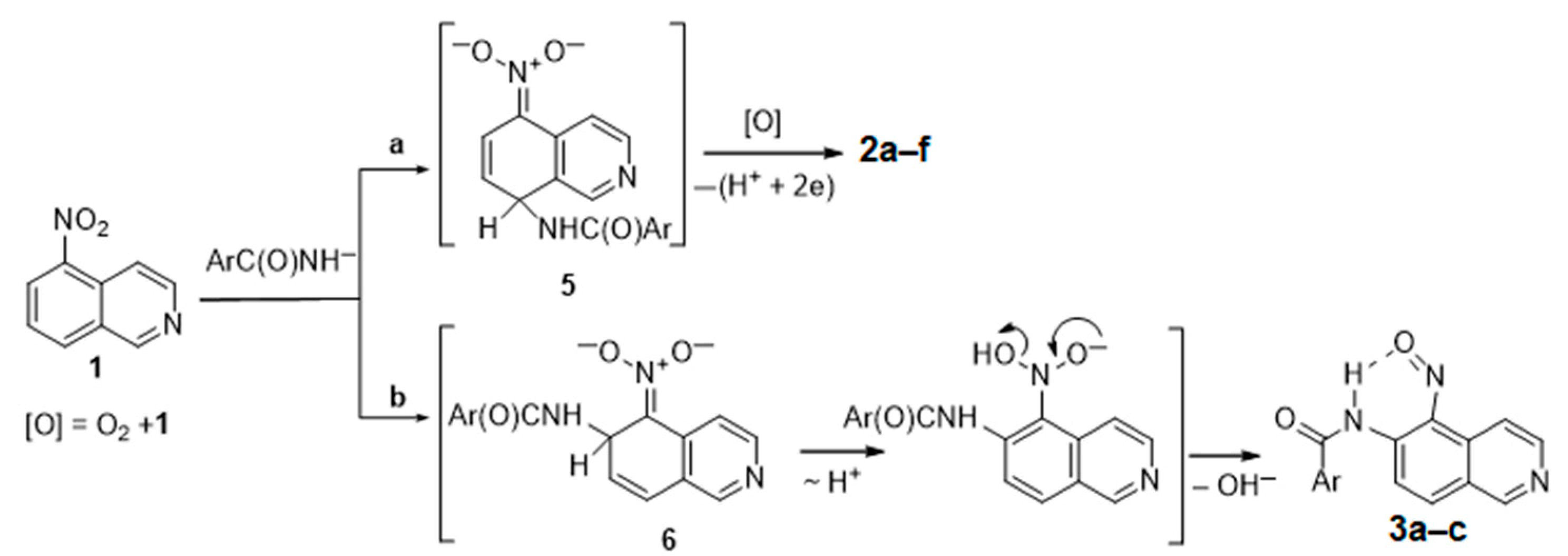{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Reaction Time, h | Excess of NaH, Equiv | Excess of Amide, Equiv | Yield, % a | |
|---|---|---|---|---|---|
| 2a | 3a | ||||
| 1 b | 1.5 | 2 | 2 | 19 | 26 |
| 2 b | 1.5 | 4 | 4 | 30 | traces |
| 3 b | 1.5 | 6 | 6 | 24 | 7.5 |
| 4 b | 1.5 | 1 | 3 | 45 | traces |
| 5 b | 1.5 | 4 | 2 | 28 | 15 |
| 6 b,c | 1.5 | 2 | 2 | 28 | traces |
| 7 d | 0.5 | 2 | 2 | 40 | traces |
| 8 b,e | 1.5 | 2 | 2 | 15 | 23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avakyan, E.K.; Borovleva, A.A.; Pobedinskaya, D.Y.; Demidov, O.P.; Ermolenko, A.P.; Larin, A.N.; Borovlev, I.V. SNH Amidation of 5-Nitroisoquinoline: Access to Nitro- and Nitroso Derivatives of Amides and Ureas on the Basis of Isoquinoline. Molecules 2022, 27, 7862. https://doi.org/10.3390/molecules27227862
Avakyan EK, Borovleva AA, Pobedinskaya DY, Demidov OP, Ermolenko AP, Larin AN, Borovlev IV. SNH Amidation of 5-Nitroisoquinoline: Access to Nitro- and Nitroso Derivatives of Amides and Ureas on the Basis of Isoquinoline. Molecules. 2022; 27(22):7862. https://doi.org/10.3390/molecules27227862
Chicago/Turabian StyleAvakyan, Elena K., Anastasia A. Borovleva, Diana Yu. Pobedinskaya, Oleg P. Demidov, Artem P. Ermolenko, Alexander N. Larin, and Ivan V. Borovlev. 2022. "SNH Amidation of 5-Nitroisoquinoline: Access to Nitro- and Nitroso Derivatives of Amides and Ureas on the Basis of Isoquinoline" Molecules 27, no. 22: 7862. https://doi.org/10.3390/molecules27227862