
Toughening Polyamidoamine Hydrogels through Covalent Grafting of Short Silk Fibers

 , , , ,
, , , ,  and
and
Abstract
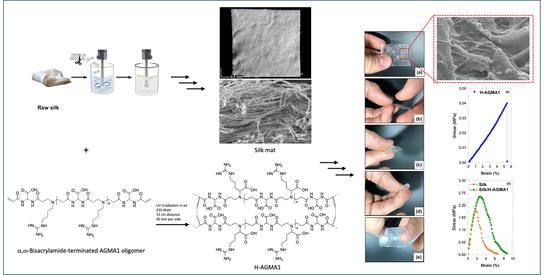
:
1. Introduction
2. Results
2.1. Rationale
2.2. Synthesis of Plain AGMA1 Hydrogels (H-AGMA1)
2.3. Synthesis of Silk/AGMA1 Composite Hydrogels (Silk/H-AGMA1)
2.4. Morphological Characterization of Silk/H-AGMA1 Composite Hydrogels
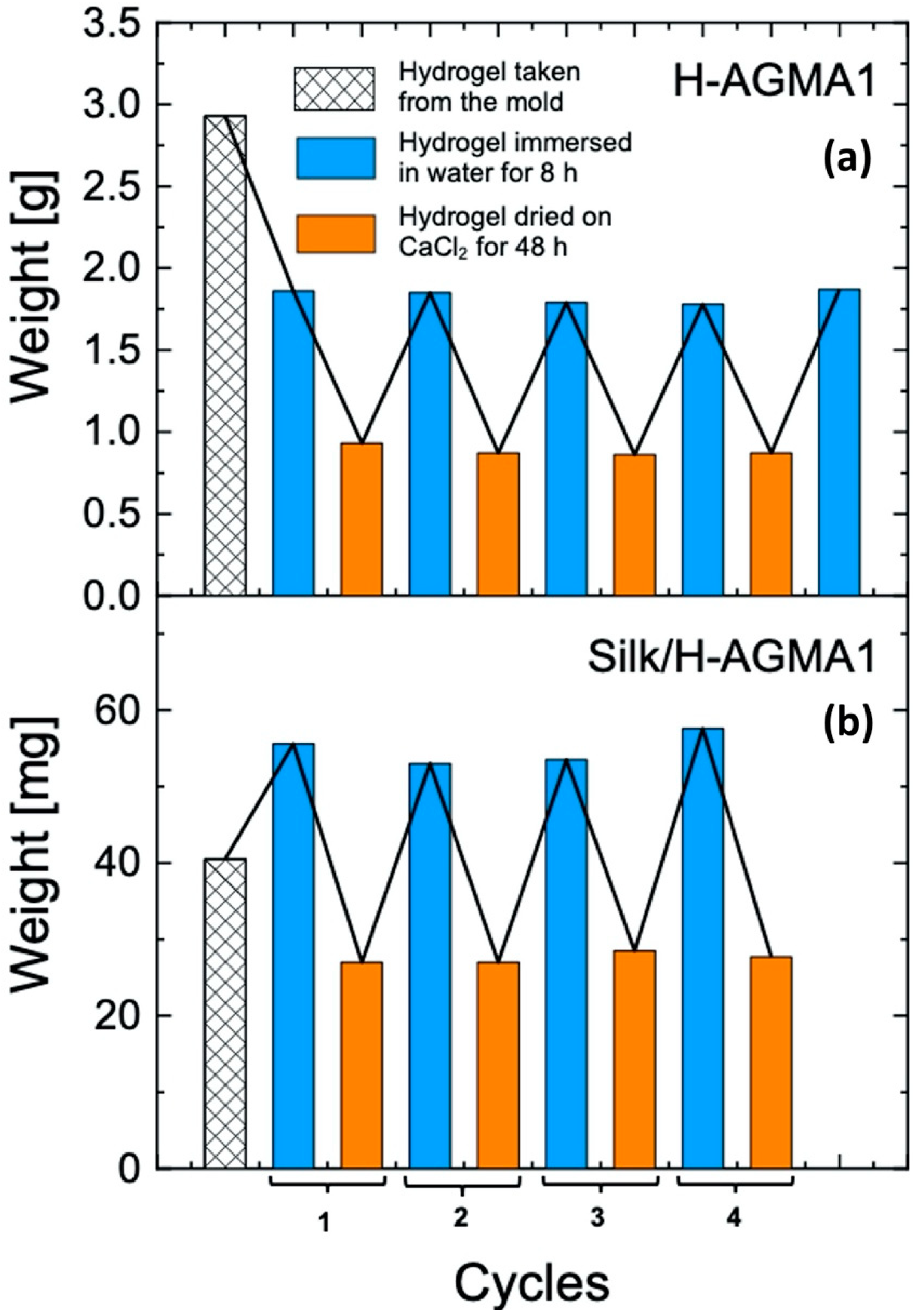
2.5. Water Uptake of H-AGMA1 and Silk/H-AGMA1 Hydrogels
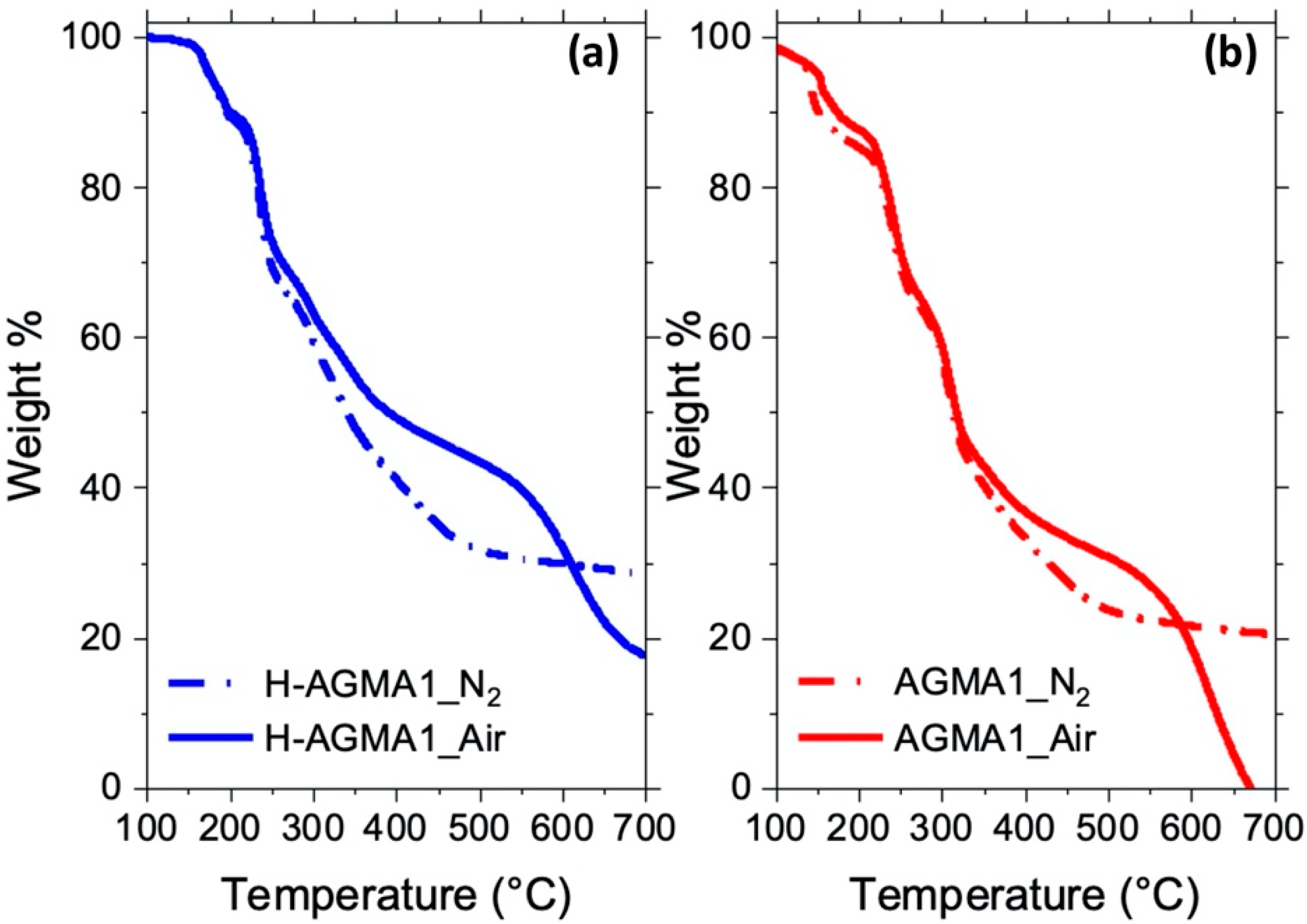
2.6. Thermal Stability of the Silk/H-AGMA1 Composite Hydrogels
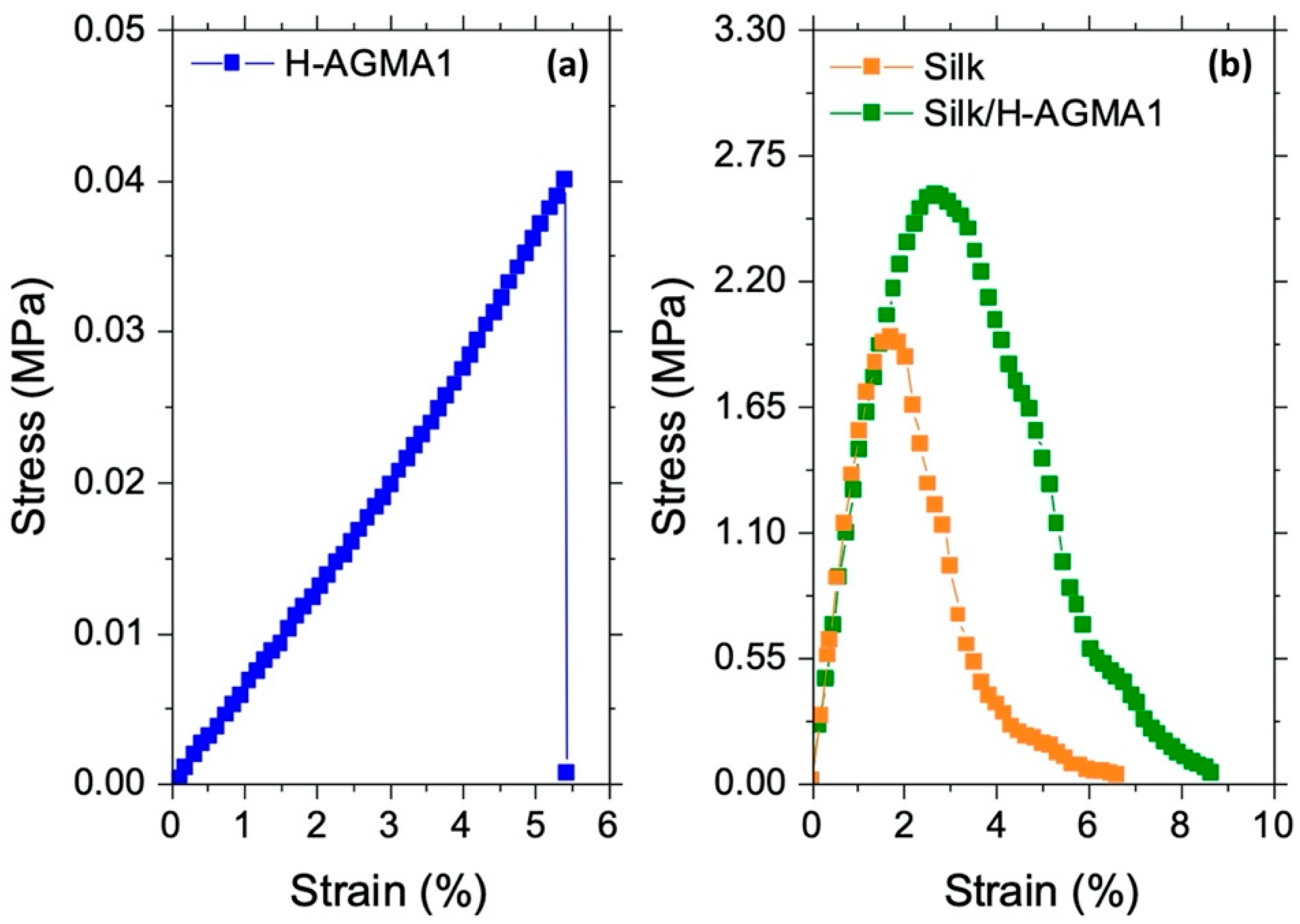
2.7. Tensile Properties of Silk/H-AGMA1 Hydrogels in the Swollen State
3. Materials and Methods
3.1. Preparation of the Silk Mat
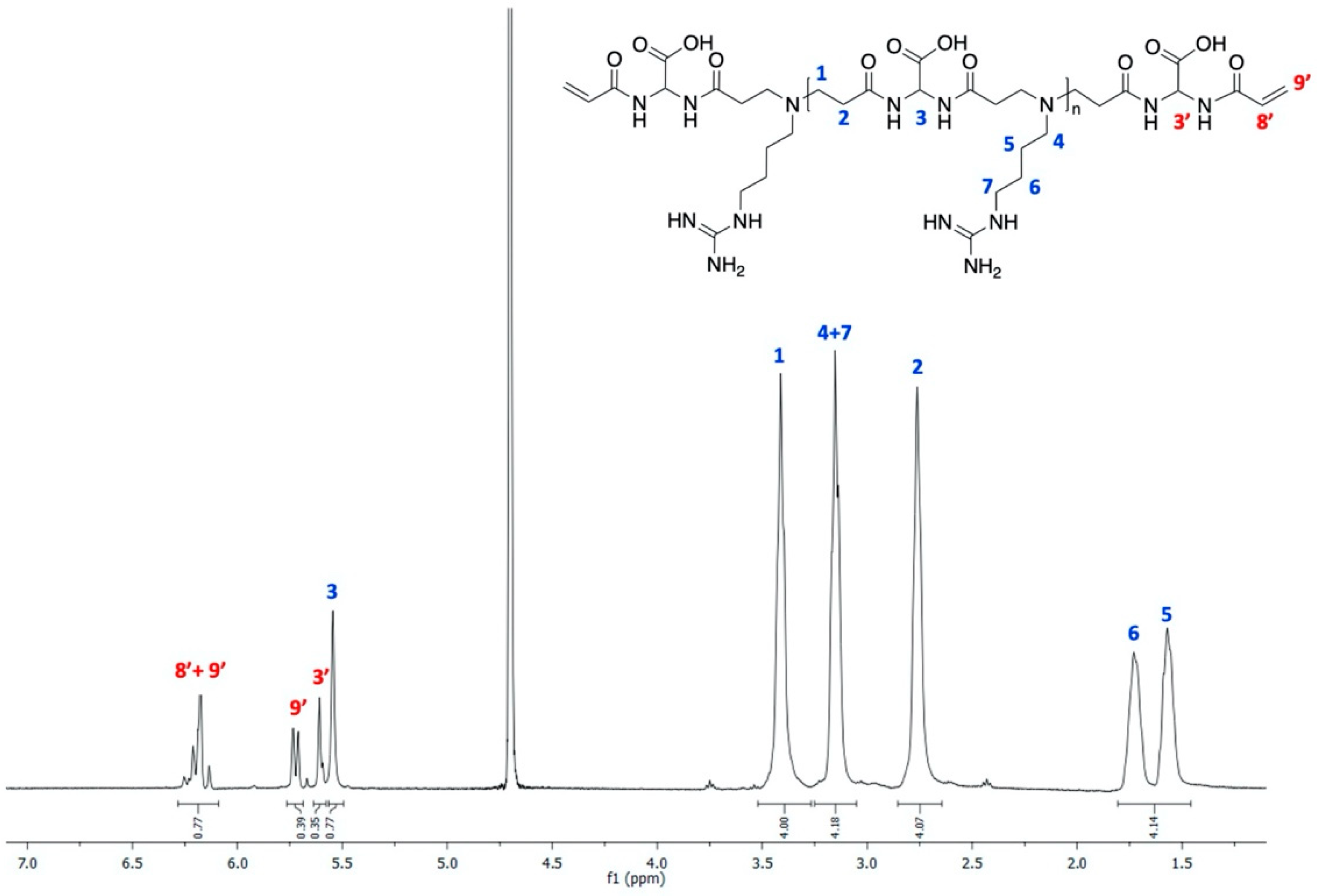
3.2. Synthesis of the α,ω-Bisacrylamide-Terminated AGMA1 Oligomer
3.3. Preparation of H-AGMA1 Hydrogel Samples
3.4. Synthesis of Silk/AGMA1 Composite Hydrogels (Silk/H-AGMA1)
3.5. Water Uptake Tests
3.6. Thermogravimetric Analysis
3.7. Scanning Electron Microscopy
3.8. Tensile Tests
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, E.M. Hydrogels, preparation, characterization and applications: A review. J. Adv. Res. 2015, 6, 105–121. [Google Scholar] [CrossRef] [Green Version]
- Sikdar, P.; Uddin, M.M.; Dip, T.M.; Islam, S.; Hoque, M.S.; Dhar, A.K.; Wu, S. Recent advances in the synthesis of smart hydrogels. Mater. Adv. 2021, 2, 4532–4573. [Google Scholar] [CrossRef]
- Mantha, S.; Pillai, S.; Khayambashi, P.; Upadhyay, A.; Zhang, Y.; Tao, O.; Pham, H.M.; Tran, S.D. Smart hydrogels in tissue engineering and regenerative medicine. Materials 2019, 12, 3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Sun, Q.; Li, Q.; Kawazoe, N.; Chen, G. Functional hydrogels with tunable structures and properties for tissue engineering applications. Front. Chem. 2018, 6, 499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.Y.; Mooney, D.J. Hydrogels for tissue engineering. Chem. Rev. 2001, 101, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P. Polyamidomines: Past, present and future perspectives. J. Polym. Sci. A: Polym. Chem. 2013, 51, 2319–2353. [Google Scholar] [CrossRef]
- Ferruti, P.; Ranucci, E.; Trotta, F.; Gianasi, E.; Evagorou, E.G.; Wasil, M.; Wilson, G.; Duncan, R. Synthesis, characterisation and antitumour activity of platinum (II) complexes of novel functionalised poly(amido amine)s. Macromol. Chem. Phys. 1999, 200, 1644–1654. [Google Scholar] [CrossRef]
- Mauro, N.; Manfredi, A.; Ranucci, E.; Procacci, P.; Laus, M.; Antonioli, D.; Mantovani, C.; Magnaghi, V.; Ferruti, P. Degradable Poly(amidoamine) Hydrogels as Scaffolds for In Vitro Culturing of Peripheral Nervous System Cells. Macromol. Biosci. 2013, 13, 332–347. [Google Scholar] [CrossRef]
- Ferruti, P.; Bianchi, S.; Ranucci, E.; Chiellini, F.; Caruso, V. Novel poly(amido-amine)-based hydrogels as scaffolds for tissue engineering. Macromol. Biosci. 2005, 5, 613–622. [Google Scholar] [CrossRef]
- Franchini, J.; Ranucci, E.; Ferruti, P.; Rossi, M.; Cavalli, R. Synthesis, Physicochemical Properties, and Preliminary Biological Characterizations of a Novel Amphoteric Agmatine-Based Poly(amidoamine) with RGD-Like Repeating Units. Biomacromolecules 2006, 7, 1215–1222. [Google Scholar] [CrossRef]
- Cavalli, R.; Bisazza, A.; Sessa, R.; Luca, P.; Fenili, F.; Manfredi, A.; Ranucci, E.; Ferruti, P. Amphoteric Agmatine Containing Polyamidoamines as Carriers for Plasmid DNA In Vitro and In Vivo Delivery. Biomacromolecules 2010, 11, 2667–2674. [Google Scholar] [CrossRef]
- Donalisio, M.; Quaranta, P.; Chiuppesi, F.; Pistello, M.; Cagno, V.; Cavalli, R.; Volante, M.; Bugatti, A.; Rusnati, M.; Ranucci, E.; et al. The AGMA1 poly(amidoamine) inhibits the infectivity of herpes simplex virus in cell lines, in human cervicovaginal histocultures, and in vaginally infected mice. Biomaterials 2016, 85, 40–53. [Google Scholar] [CrossRef]
- Tonna, N.; Bianco, F.; Matteoli, M.; Cagnoli, C.; Antonucci, F.; Manfredi, A.; Mauro, N.; Ranucci, E.; Ferruti, P. A soluble biocompatible guanidine-containing polyamidoamine as promoter of primary brain cell adhesion and in vitro cell culturing. Sci. Technol. Adv. Mater. 2014, 15, 045007. [Google Scholar] [CrossRef] [PubMed]
- Ferruti, P.; Bianchi, S.; Ranucci, E.; Chiellini, F.; Piras, A.M. Novel Agmatine-Containing Poly(amidoamine) Hydrogels as Scaffolds for Tissue Engineering. Biomacromolecules 2005, 6, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, G.; Fenili, F.; Gianfelice, A.; Bongiorno, G.; Marchesi, D.; Scopelliti, P.E.; Borgonovo, A.; Podestà, A.; Indrieri, M.; Ranucci, E.; et al. Direct Microfabrication of Topographical and Chemical Cues for the Guided Growth of Neural Cell Networks on Polyamidoamine Hydrogels. Macromol. Biosci. 2010, 10, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Jacchetti, E.; Emilitri, E.; Rodighiero, S.; Indrieri, M.; Gianfelice, A.; Lenardi, C.; Podestà, A.; Ranucci, E.; Ferruti, P.; Milani, P. Biomimetic poly(amidoamine) hydrogels as synthetic materials for cell culture. J. Nanobiotechnol. 2008, 6, 14. [Google Scholar] [CrossRef] [Green Version]
- Rossi, E.; Gerges, I.; Tocchio, A.; Tamplenizza, M.; Aprile, P.; Recordati, C.; Martello, F.; Martin, I.; Milani, P.; Lenardi, C. Biologically and mechanically driven design of an RGD-mimetic macroporous foam for adipose tissue engineering applications. Biomaterials 2016, 104, 65–77. [Google Scholar] [CrossRef]
- Magnaghi, V.; Conte, V.; Procacci, P.; Pivato, G.; Cortese, P.; Cavalli, E.; Pajardi, G.; Ranucci, E.; Fenili, F.; Manfredi, A.; et al. Biological performance of a novel biodegradable polyamidoamine hydrogel as guide for peripheral nerve regeneration. J. Biomed. Mater. Res. A 2011, 98, 19–30. [Google Scholar] [CrossRef]
- Mauro, N.; Chiellini, F.; Bartoli, C.; Gazzarri, M.; Laus, M.; Antonioli, D.; Griffiths, P.; Manfredi, A.; Ranucci, E.; Ferruti, P. RGD-mimic polyamidoamine–montmorillonite composites with tunable stiffness as scaffolds for bone tissue-engineering applications. J. Tissue Eng. Regen. Med. 2017, 11, 2164–2175. [Google Scholar] [CrossRef]
- Li, J.; Gao, L.; Xu, R.; Ma, S.; Ma, Z.; Liu, Y.; Wu, Y.; Feng, L.; Cai, M.; Zhou, F. Fibers reinforced composite hydrogels with improved lubrication and load-bearing capacity. Friction 2022, 10, 54–67. [Google Scholar] [CrossRef]
- Huang, Y.; King, D.R.; Sun, T.L.; Nonoyama, T.; Kurokawa, T.; Nakajima, T.; Gong, J.P. Energy-dissipative matrices enable synergistic toughening in fiber reinforced soft composites. Adv. Funct. Mater. 2017, 27, 1605350. [Google Scholar] [CrossRef] [Green Version]
- Liao, I.-C.; Moutos, F.T.; Estes, B.T.; Zhao, X.; Guilak, F. Composite three-dimensional woven scaffolds with interpenetrating network hydrogels to create functional synthetic articular cartilage. Adv. Funct. Mater. 2013, 23, 5833–5839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Shi, Z.; Chen, X.; Yang, G.; Lenardi, C.; Liu, C. Microstructural and mechanical characteristics of PHEMA-based nanofibre-reinforced hydrogel under compression. Composites Part B 2015, 76, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Deng, L.; Zhang, J.; Yin, L.; Dong, A. Composites of electrospun-fibers and hydrogels: A potential solution to current challenges in biological and biomedical field. J. Biomed. Mater. Res. Part B 2016, 104B, 640–656. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.O.; Antunes, J.C.; Felgueiras, H.P. Recent advances in fiber–hydrogel composites for wound healing and drug delivery systems. Antibiotics 2021, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Cao, C.; Wang, Q.; Gonzalez, M.; Dolbow, J.E.; Zhao, X. Design of stiff, tough and stretchy hydrogel composites via nanoscale hybrid crosslinking and macroscale fiber reinforcement. Soft Matter 2014, 10, 7519–7527. [Google Scholar] [CrossRef]
- Gualandi, C.; Bloise, N.; Mauro, N.; Ferruti, P.; Manfredi, A.; Sampaolesi, M.; Liguori, A.; Laurita, R.; Gherardi, M.; Colombo, M.; et al. Poly-L-Lactic Acid Nanofiber-Polyamidoamine Hydrogel Composites: Preparation, Properties, and Preliminary Evaluation as Scaffolds for Human Pluripotent Stem Cell Culturing. Macromol. Biosci. 2016, 16, 1533–1544. [Google Scholar] [CrossRef]
- Vepari, C.; Kaplan, D.L. Silk as a biomaterial. Prog. Polym. Sci. 2007, 32, 991–1007. [Google Scholar] [CrossRef]
- Holland, C.; Numata, K.; Rnjak-Kovacina, J.; Seib, F.P. The biomedical use of silk: Past, present, future. Adv. Healthcare Mater. 2019, 8, 1800465. [Google Scholar] [CrossRef] [Green Version]
- Rockwood, D.N.; Preda, R.C.; Yücel, T.; Wang, X.; Lovett, M.L.; Kaplan, D.L. Materials fabrication from Bombyx mori silk fibroin. Nat. Protoc. 2011, 6, 1612–1631. [Google Scholar] [CrossRef] [Green Version]
- Byram, P.K.; Das, L.; Sunka, K.C.; Kulkarni, G.; Dhara, S.; Chakravorty, N. Silk Fibroin-Based Biomaterials in Biomedical Applications. In Functional Biomaterials; Jana, S., Jana, S., Eds.; Springer: Singapore, Singapore, 2022; pp. 203–244. [Google Scholar]
- Gholipourmalekabadi, M.; Sapru, S.; Samadikuchaksaraei, A.; Reis, R.L.; Kaplan, D.L.; Kundu, S.C. Silk fibroin for skin injury repair: Where do things stand? Adv. Drug Delivery Rev. 2020, 153, 28–53. [Google Scholar] [CrossRef] [PubMed]
- Font Tellado, S.; Chiera, S.; Bonani, W.; Poh, P.S.P.; Migliaresi, C.; Motta, A.; Balmayor, E.R.; van Griensven, M. Heparin functionalization increases retention of TGF-β2 and GDF5 on biphasic silk fibroin scaffolds for tendon/ligament-to-bone tissue engineering. Acta Biomater. 2018, 72, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Gambari, L.; Amore, E.; Raggio, R.; Bonani, W.; Barone, M.; Lisignoli, G.; Grigolo, B.; Motta, A.; Grassi, F. Hydrogen sulfide-releasing silk fibroin scaffold for bone tissue engineering. Mater. Sci. Eng. C 2019, 102, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Khafaji, M.; Naji, M.; Vossoughi, M.; Alemzadeh, I.; Haghighipour, N. A Biomimetic Heparinized Composite Silk-Based Vascular Scaffold with sustained Antithrombogenicity. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Flory, P. Molecular Size and Chemical Reactivity; Principles of Condensation Polymerization. In Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953; pp. 91–95. [Google Scholar]
- Samie, M.; Muhammad, N.; Arfat Yameen, M.; Anwar Chaudhry, A.; Khalid, H.; Khan, A.F. Aqueous Solution of a Basic Ionic Liquid: A Perspective Solvent for Extraction and Regeneration of Silk Powder from Bombyx mori Silk Cocoons. J. Polym. Environ. 2020, 28, 657–667. [Google Scholar] [CrossRef]
- Malucelli, G. Biomacromolecules and Bio-Sourced Products for the Design of Flame Retarded Fabrics: Current State of the Art and Future Perspectives. Molecules 2019, 24, 3774. [Google Scholar] [CrossRef] [Green Version]
- Vandersall, H.J. Intumescent coating system, their development and chemistry. J. Fire Flam. 1971, 2, 97–140. [Google Scholar]
- Forte, C.; Alongi, J.; Beduini, A.; Borsacchi, S.; Calucci, L.; Carosio, F.; Ferruti, P.; Ranucci, E. The thermo-oxidative behavior of cotton coated with an intumescent flame retardant glycine-derived polyamidoamine: A multi-technique study. Polymers 2021, 13, 4382. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Tonset10% a [°C] | Tmax1 b [°C] | Tmax2 c [°C] | Tmax3 d [°C] | Residual Mass Fraction at 700 °C [%] |
|---|---|---|---|---|---|
| Nitrogen | |||||
| H-AGMA1 | 197 | 172/194 | 235 | 301 | 28 |
| Silk | 302 | - | - | 319/390 | 29 |
| Silk/H-AGMA1 composite | 251 | 239 | 307/414 | 31 | |
| Air | |||||
| H-AGMA1 | 201 | 165/194 | 236 | 295 | 18 |
| Silk | 290 | - | - | 320 | 0 |
| Silk/H-AGMA1 composite | 253 | - | 242 | 324 | 0 |
| Sample | Length (mm) | Width (mm) | Thickness (mm) | Elastic Modulus (MPa) | Maximum Stress (MPa) | Ultimate Tensile Strength (N) | Elongation at Break (%) |
|---|---|---|---|---|---|---|---|
| H-AGMA1 | 30 ± 0 | 6 ± 0 | 0.55 ± 0.06 | 0.3 ± 0.1 | 0.04 ± 0.01 | 0.10 ± 0.06 | 5.3 ± 3.1 |
| Silk | 40.6 ± 1.9 | 6.7 ± 1.0 | 0.21 ± 0 | 192.7 ± 55.4 | 1.96 ± 0.60 | 2.79 ± 0.43 | 6.8 ± 2.1 |
| Silk/H-AGMA1 | 34 ± 0.12 | 7.2 ± 0.5 | 0.13 ± 0.01 | 162.7 ± 17.8 | 2.58 ± 0.87 | 1.95 ± 0.62 | 8.2 ± 1.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maggi, F.; Manfredi, A.; Carosio, F.; Maddalena, L.; Alongi, J.; Ferruti, P.; Ranucci, E. Toughening Polyamidoamine Hydrogels through Covalent Grafting of Short Silk Fibers. Molecules 2022, 27, 7808. https://doi.org/10.3390/molecules27227808
Maggi F, Manfredi A, Carosio F, Maddalena L, Alongi J, Ferruti P, Ranucci E. Toughening Polyamidoamine Hydrogels through Covalent Grafting of Short Silk Fibers. Molecules. 2022; 27(22):7808. https://doi.org/10.3390/molecules27227808
Chicago/Turabian StyleMaggi, Filippo, Amedea Manfredi, Federico Carosio, Lorenza Maddalena, Jenny Alongi, Paolo Ferruti, and Elisabetta Ranucci. 2022. "Toughening Polyamidoamine Hydrogels through Covalent Grafting of Short Silk Fibers" Molecules 27, no. 22: 7808. https://doi.org/10.3390/molecules27227808