

Eight Indole Alkaloids from the Roots of Maerua siamensis and Their Nitric Oxide Inhibitory Effects
 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
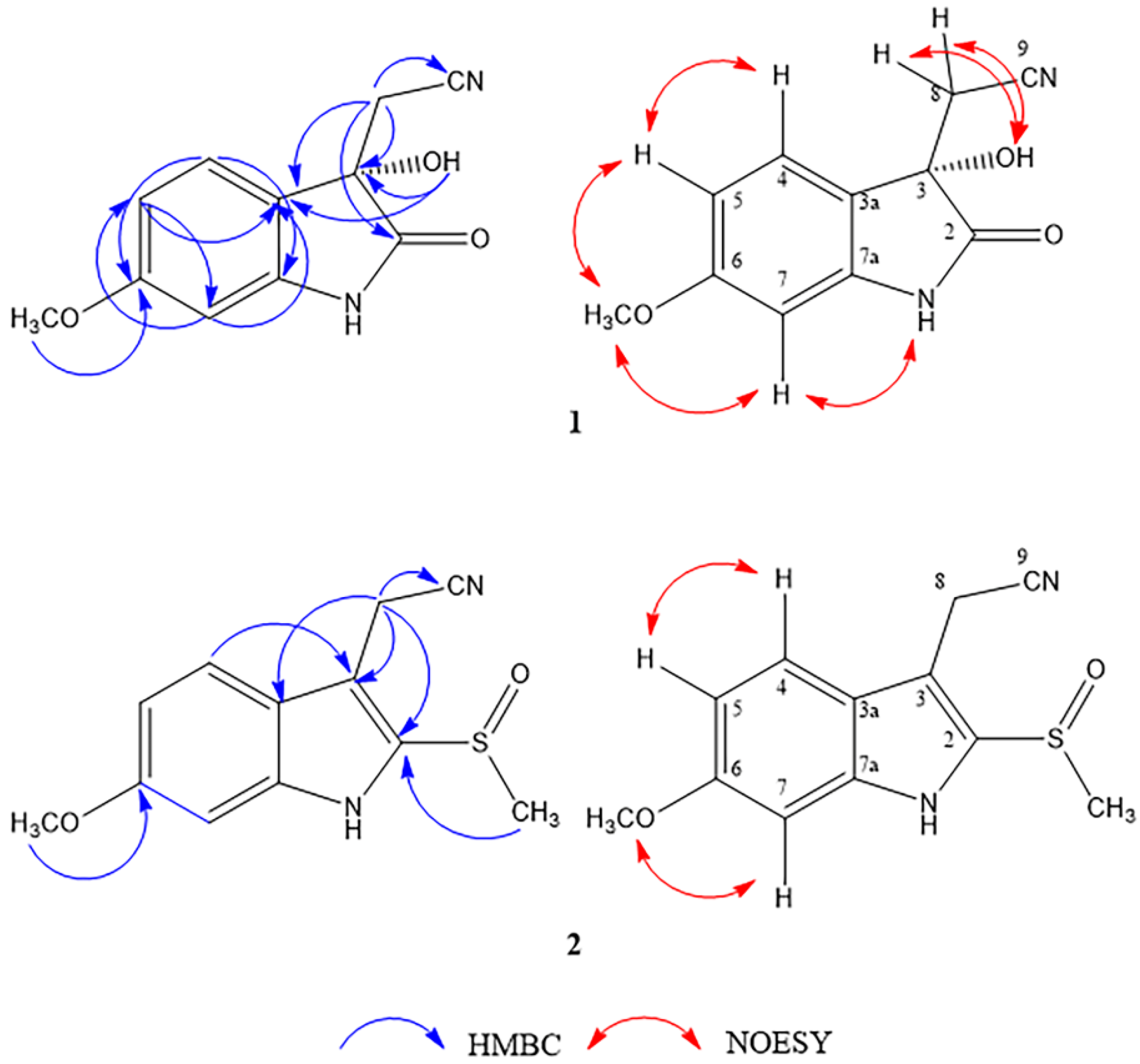
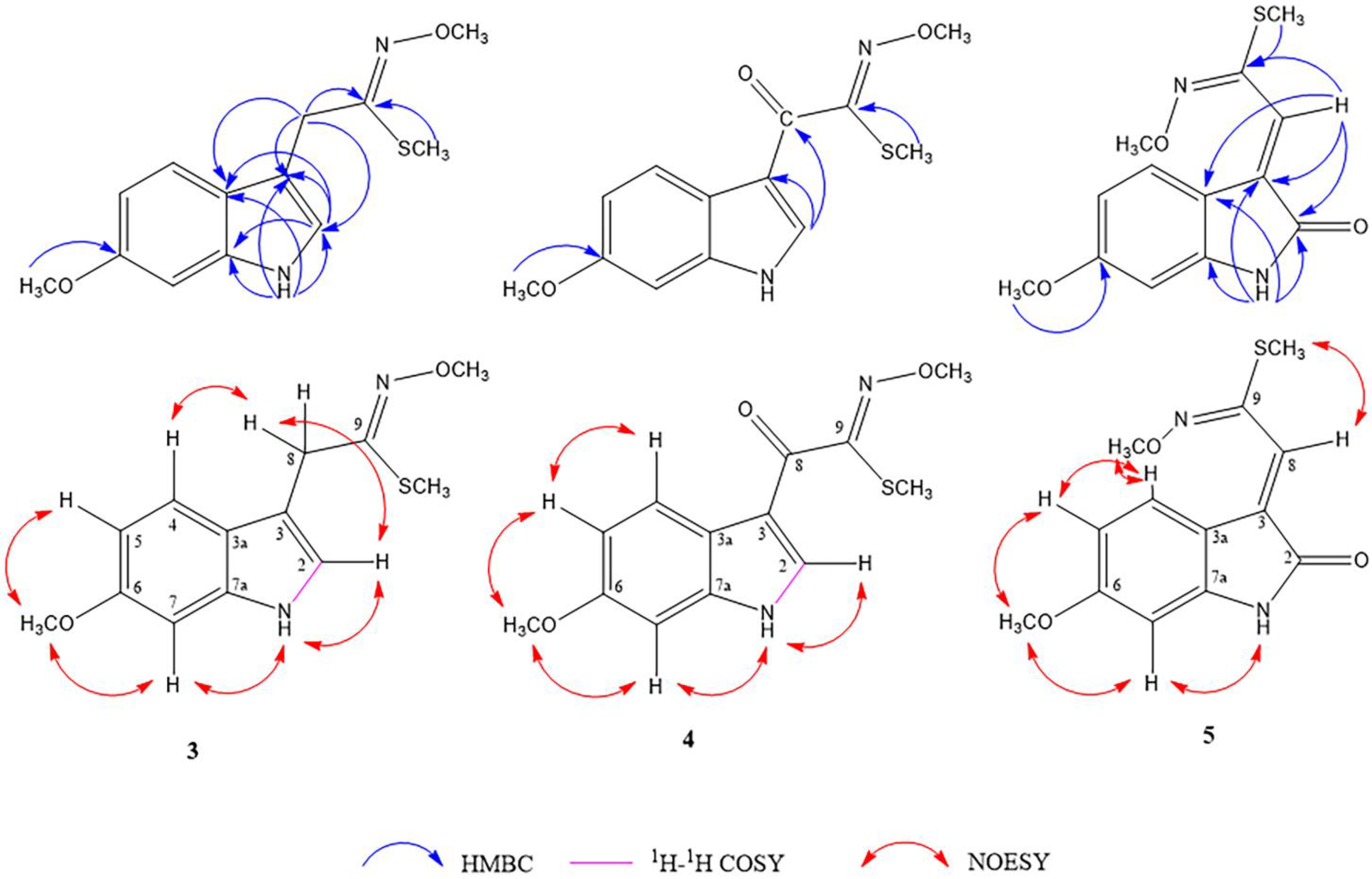
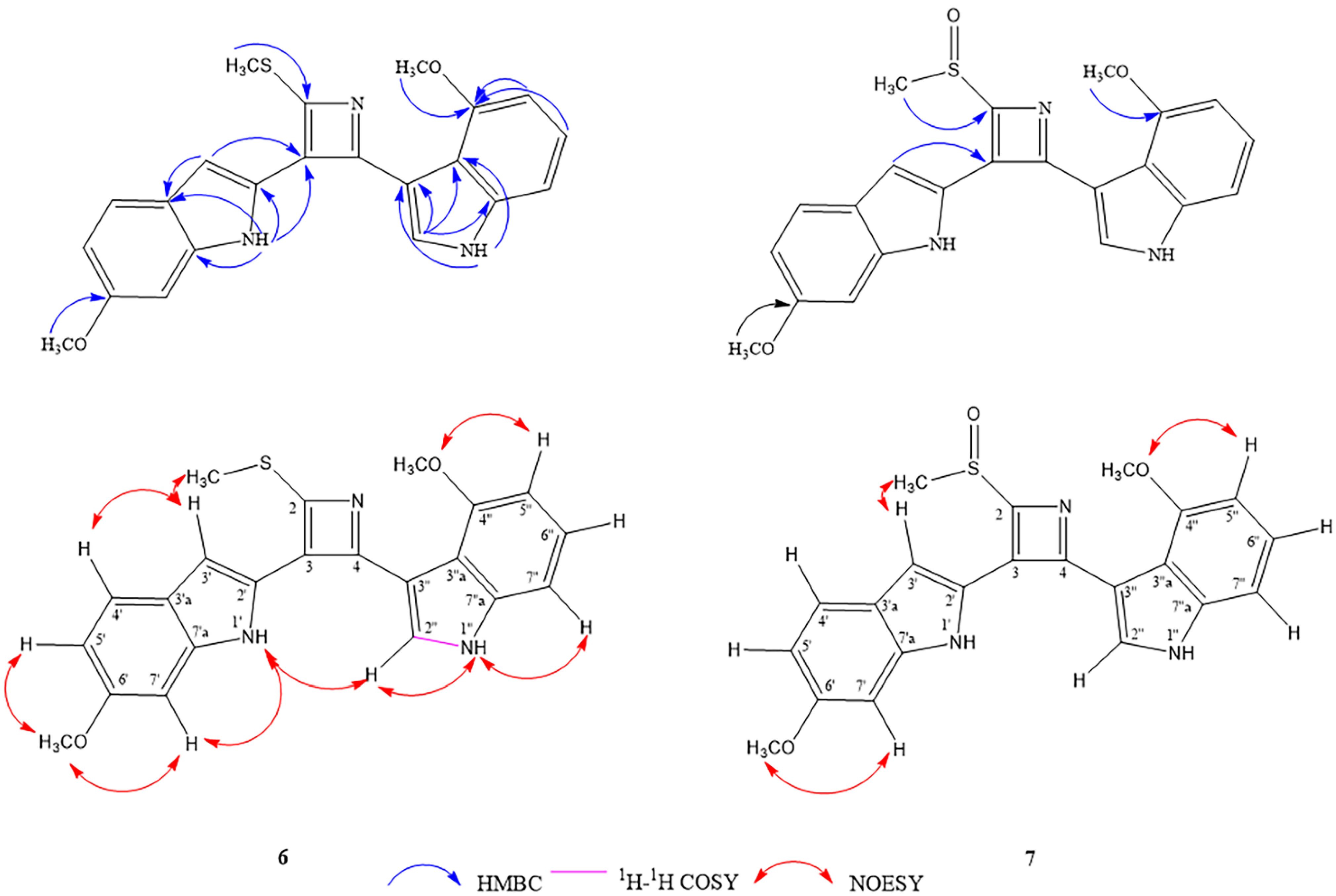
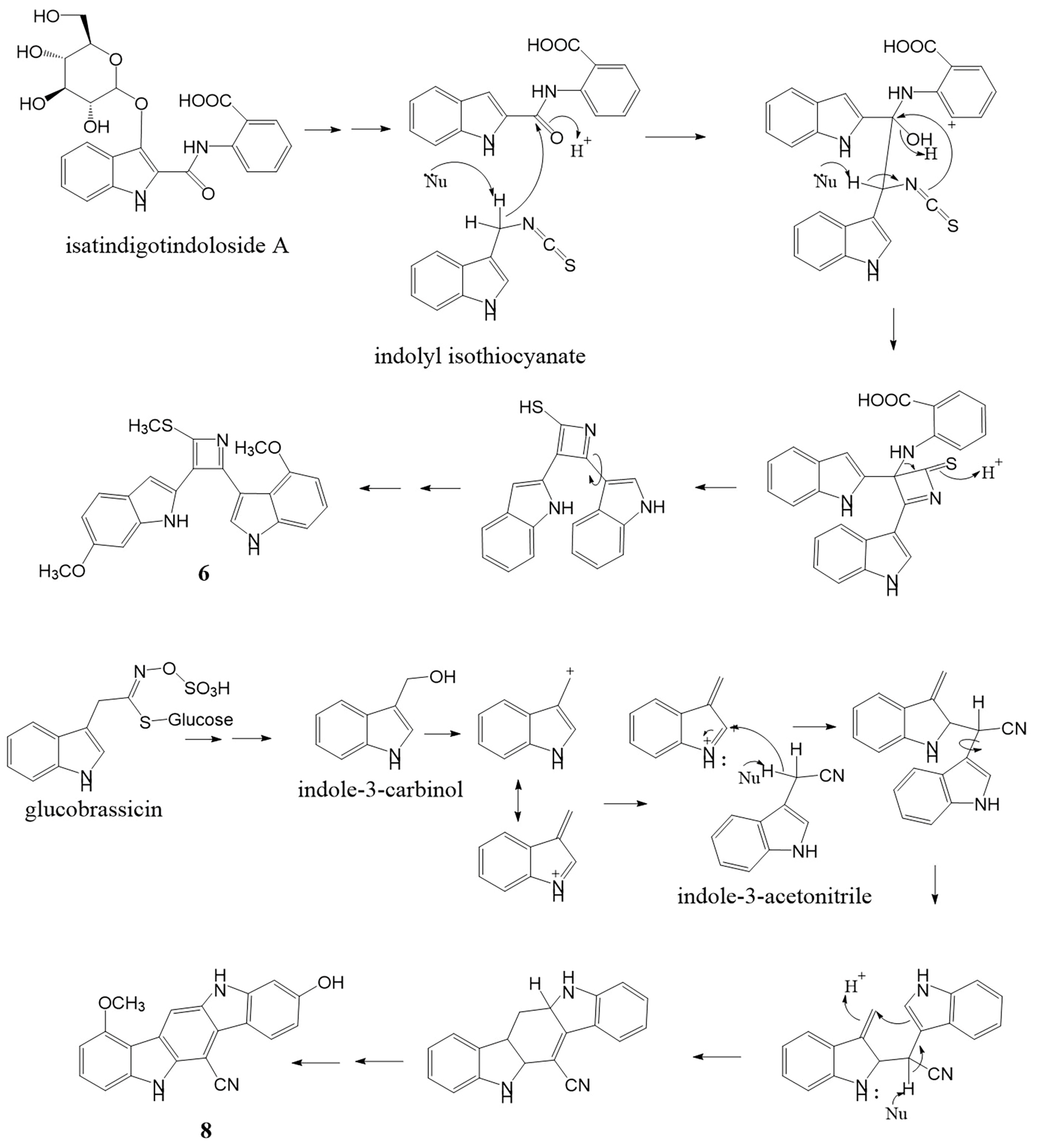
2.1. Structure Determination of Isolated Compounds
2.2. Computational Analysis
2.3. Inhibition of Nitric Oxide Production of Isolated Compounds
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Computational Detail
3.5. Inhibitory Activity of NO Production
3.5.1. Materials
3.5.2. Cell Culture
3.5.3. Preparation of Test Solutions
3.5.4. Stimulation of Inflammation in Raw264.7 Cells
3.5.5. Measurement of NO Production
3.5.6. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
Sample Availability
References
- Royal Botanic Gardens, Kew. Plants of the World Online. Available online: https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:5968-1 (accessed on 26 August 2022).
- Stevenson, P.C.; Green, P.W.C.; Farrell, I.W.; Brankin, A.; Mvumi, B.M.; Belmain, S.R. Novel agmatine derivatives in Maerua edulis with bioactivity against Callosobruchus maculatus, a cosmopolitan storage insect pest. Front. Plant Sci. 2018, 9, 1506. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mogib, M. A lupane triterpenoid from Maerua oblongifolia. Phytochemistry 1999, 51, 445–448. [Google Scholar] [CrossRef]
- Ibraheim, Z.Z. A new ionol glucoside from Maerua crassifolia Forssk grown in Egypt. Bull. Pharm. Sci. Assiut Univ. 1995, 18, 27–31. [Google Scholar] [CrossRef]
- Akuodor, G.C.; Essien, A.D.; Akpan, J.L.; Chilaka, K.C.; Jane, N.; Uwaezuoke, I.; Nwadike, K.I.; Nwobodo, N.N.; Ezeokpo, B.C. Analgesic, anti-inflammatory and antipyretic activities of methanolic leaf extract of Maerua crassifolia. J. Coast. Life Med. 2016, 4, 225–230. [Google Scholar] [CrossRef]
- Hiben, M.G.; de Haan, L.; Spenkelink, B.; Wesseling, S.; Vervoort, J.; Rietjens, I.M.C.M. Induction of peroxisome proliferator activated receptor γ (PPARγ) mediated gene expression and inhibition of induced nitric oxide production by Maerua subcordata (Gilg) DeWolf. BMC Complement. Med. Ther. 2020, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Medicines Regulation Division. Ministerial Notification (B.E. 2556) (2013) on Traditional Nonprescription Drugs. Available online: https://www.fda.moph.go.th/sites/drug/Shared%20Documents/Law03-TheMinistryOfHealth/Law03-07-03.pdf (accessed on 26 August 2022).
- The Forest Herbarium. Tem Smitinand’s Thai Plant Names; Revised Edition 2011; Prachachon: Bangkok, Thailand, 2011; p. 339. [Google Scholar]
- Theanphong, O.; Somwong, P. Combination of selected Thai traditional pain relief medicinal plants with anti-inflammatory abilities in a protein denaturation assay. Pharmacia 2022, 69, 745. [Google Scholar] [CrossRef]
- Nobsathian, S.; Bullangpoti, V.; Kumrungsee, N.; Wongsa, N.; Ruttanakum, D. Larvicidal effect of compounds isolated from Maerua siamensis (Capparidaceae) against Aedes aegypti (Diptera: Culicidae) larvae. Chem. Biol. Technol. Agric. 2018, 5, 8. [Google Scholar] [CrossRef]
- Hall, J.C.; Sytsma, K.J.; Iltis, H.H. Phylogeny of Capparaceae and Brassicaceae based on chloroplast sequence data. Am. J. Bot. 2002, 89, 1826–1842. [Google Scholar] [CrossRef]
- Yang, L.; Wang, G.; Wang, M.; Jiang, H.; Chen, L.; Zhao, F.; Qiu, F. Indole alkaloids from the roots of Isatis indigotica and their inhibitory effects on nitric oxide production. Fitoterapia 2014, 95, 175–181. [Google Scholar] [CrossRef]
- Kinashi, H.; Suzuki, Y.; Takeuchi, S.; Kawarada, A. Possible metabolic intermediates from IAA to β-Acid in rice bran. Agric. Biol. Chem. 1976, 40, 2465–2470. [Google Scholar]
- Gao, Z.-H.; Kong, L.-M.; Zou, X.-S.; Shi, Y.-M.; Shang, S.-Z.; Luo, H.-R.; Liang, C.-Q.; Li, X.-N.; Li, Y.; Du, X.; et al. Four new indole alkaloids from Plantago asiatica. Nat. Prod. Bioprospect. 2012, 2, 249–254. [Google Scholar] [CrossRef]
- Chen, M.; Gan, L.; Lin, S.; Wang, X.; Li, L.; Li, Y.; Zhu, C.; Wang, Y.; Jiang, B.; Jiang, J.; et al. Alkaloids from the root of Isatis indigotica. J. Nat. Prod. 2012, 75, 1167–1176. [Google Scholar] [CrossRef]
- Faisal, S.; Basha, A.F.; Siddiqui, H.; Basha, F.Z. O-Alkylation of menthone oxime: Synthesis and 13C NMR studies of a series of novel oxime ethers. Synth. Commun. 2010, 40, 3101–3108. [Google Scholar] [CrossRef]
- Czerniawski, P.; Bednarek, P. Glutathione-S-transferases in the biosynthesis of sulfur-containing secondary metabolites in Brassicaceae plants. Front. Plant Sci. 2018, 9, 1639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ruan, D.; Li, J.; Chen, Z.; Zhu, W.; Guo, F.; Chen, K.; Li, Y.; Wang, R. Four undescribed sulfur-containing indole alkaloids with nitric oxide inhibitory activities from Isatis tinctoria L. roots. Phytochemistry 2020, 174, 112337. [Google Scholar] [CrossRef]
- Wahlström, N.; Slätt, J.; Stensland, B.; Ertan, A.; Bergman, J.; Janosik, T. Synthetic applications of cyanoacetylated bisindoles: Synthesis of novel cycloheptadiindoles, indolocarbazoles, and related aza analogues. J. Org. Chem. 2007, 72, 5886–5889. [Google Scholar] [CrossRef]
- Faust, D.; Nikolova, T.; Wätjen, W.; Kaina, B.; Dietrich, C. The Brassica-derived phytochemical indolo[3,2-b]carbazole protects against oxidative DNA damage by aryl hydrocarbon receptor activation. Arch. Toxicol. 2017, 91, 967–982. [Google Scholar] [CrossRef]
- Koper, J.E.B.; Kortekaas, M.; Loonen, L.M.P.; Huang, Z.; Wells, J.M.; Gill, C.I.R.; Pourshahidi, L.K.; McDougall, G.; Rowland, I.; Pereira-Caro, G.; et al. Aryl hydrocarbon receptor activation during in vitro and in vivo digestion of raw and cooked broccoli (Brassica oleracea var. italica). Food Funct. 2020, 11, 4026–4037. [Google Scholar] [CrossRef]
- Wallace, J.L. Nitric oxide as a regulator of inflammatory processes. Mem. Inst. Oswaldo Cruz 2005, 100 (Suppl. 1), 5–9. [Google Scholar] [CrossRef]
- Cinelli, M.A.; Do, H.T.; Miley, G.P.; Silverman, R.B. Inducible nitric oxide synthase: Regulation, structure, and inhibition. Med. Res. Rev. 2020, 40, 158–189. [Google Scholar] [CrossRef]
- Needleman, P.; Manning, P.T. Interactions between the inducible cyclooxygenase (COX-2) and nitric oxide synthase (iNOS) pathways: Implications for therapeutic intervention in osteoarthritis. Osteoarthr. Cartil. 1999, 7, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Royal Botanic Gardens, Kew. Plants of the World Online. Available online: https://powo.science.kew.org/taxon/urn:lsid:ipni.org:names:285798-1 (accessed on 26 August 2022).
- Zhang, D.; Sun, Y.; Chen, Z.; Jia, Q.; Zhu, W.; Chen, K.; Li, Y.; Wang, R. Bisindole alkaloids with nitric oxide inhibitory activities from an alcohol extract of the Isatis indigotica roots. Fitoterapia 2020, 146, 104654. [Google Scholar] [CrossRef]
- Zhang, D.; Du, K.; Zhao, Y.; Shi, S.; Wu, Y.; Jia, Q.; Chen, K.; Li, Y.; Wang, R. Indole alkaloid glycosides from Isatis tinctoria roots. Nat. Prod. Res. 2021, 35, 244–250. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. A; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Eaknai, W.; Bunwatcharaphansakun, P.; Phungbun, C.; Jantimaporn, A.; Chaisri, S.; Boonrungsiman, S.; Nimmannit, U.; Khongkow, M. Ethanolic fenugreek extract: Its molecular mechanisms against skin aging and the enhanced functions by nanoencapsulation. Pharmaceuticals 2022, 15, 254. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, J.H.; Moon, S.H.; Ahn, D.U.; Paik, H.D. Ovalbumin hydrolysates inhibit nitric oxide production in LPS-induced RAW 264.7 macrophages. Food Sci. Anim. Resour. 2020, 40, 274–285. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δH, Multiplicity (J in Hz) a | δC | δH, Multiplicity (J in Hz) b | δC | |
| NH-1 | 9.52, br s | |||
| 2 | 178.2 | 132.5 | ||
| 3 | 73.8 | 110.8 | ||
| 3a | 122.8 | 121.4 | ||
| 4 | 7.48, d (8.4) | 126.9 | 7.63, d (9.0) | 121.4 |
| 5 | 6.63, dd (8.4, 2.4) | 108.4 | 6.87, dd (9.0, 2.4) | 113.4 |
| 6 | 163.4 | 160.6 | ||
| 7 | 6.53, d (2.4) | 98.8 | 6.98, d (2.4) | 95.3 |
| 7a | 144.7 | 140.3 | ||
| 8a 8b | 3.09, d (16.8) 2.89, d (16.8) | 28.0 | 4.18, d (18.0) 4.13, d (18.0) | 13.0 |
| 9 | 117.7 | 118.9 | ||
| 2-SOCH3 | 2.16, s | 40.4 | ||
| 3-OH | 5.44, s | |||
| 6-OCH3 | 3.81, s | 56.5 | 3.86, s | 55.9 |
| Position | 3 | 4 | 5 | |||
|---|---|---|---|---|---|---|
| δH, Multiplicity (J in Hz) a | δC | δH, Multiplicity (J in Hz) a | δC | δH, Multiplicity (J in Hz) a | δC | |
| NH-1 | 10.72, br s | 12.10, br s | 10.63, br s | |||
| 2 | 7.03, d (2.0) | 123.1 | 7.97, s | 136.7 | 168.6 | |
| 3 | 107.8 | 113.6 | 131.1 | |||
| 3a | 121.5 | 118.5 | 113.3 | |||
| 4 | 7.33, d (8.8) | 118.9 | 7.92, d (8.4) | 121.4 | 7.87, d (8.4) | 126.5 |
| 5 | 6.64, dd (8.8, 2.4) | 108.8 | 6.89, dd (8.4) | 112.3 | 6.52, dd (8.4, 2.4) | 106.8 |
| 6 | 155.6 | 156.9 | 162.2 | |||
| 7 | 6.84, d (2.4) | 94.5 | 7.00, s | 95.8 | 6.41, d (2.4) | 96.6 |
| 7a | 136.8 | 138.0 | 145.6 | |||
| 8 | 3.80, s | 25.9 | 182.5 | 6.80, s | 119.9 | |
| 9 | 157.8 | 155.9 | 150.8 | |||
| 6-OCH3 | 3.74, s | 55.2 | 3.79, s | 55.3 | 3.78, s | 55.5 |
| SCH3 | 2.17, s | 12.5 | 2.43, s | 12.7 | 2.35, s | 12.6 |
| N-OCH3 | 3.86, s | 61.5 | 3.73, s | 61.9 | 4.00, s | 62.5 |
| Position | 6 | 7 | ||
|---|---|---|---|---|
| δH, Multiplicity (J in Hz) a | δC | δH, Multiplicity (J in Hz) b | δC | |
| 2-azete | 144.8 | 151.7 | ||
| 3-azete | 133.4 | 137.8 | ||
| 4-azete | 139.5 | 142.7 | ||
| NH-1′ | 10.71, br s | |||
| 2′ | 129.3 | 130.8 | ||
| 3′ | 7.79, br s | 108.5 | 8.43, s | 109.3 |
| 3′a | 114.1 | 116.3 | ||
| 4′ | 8.05, d (8.4) | 122.6 | 8.12, d (8.4) | 123.7 |
| 5′ | 6.78, dd (8.4, 2.0) | 108.7 | 6.93, dd (8.4, 2.0) | 111.8 |
| 6′ | 160.0 | 163.0 | ||
| 7′ | 6.99, d (2.4) | 94.8 | 7.05, d (2.0) | 95.8 |
| 7′a | 142.9 | 144.9 | ||
| NH-1″ | 11.52, br s | |||
| 2″ | 7.51, d (2.0) | 125.0 | 7.54, s | 126.1 |
| 3″ | 113.0 | 113.2 | ||
| 3″a | 116.4 | 118.2 | ||
| 4″ | 153.9 | 155.6 | ||
| 5″ | 6.56, dd (6.0, 2.4) | 100.4 | 6.60, d (6.8) | 101.7 |
| 6″ | 7.10, overlapped | 122.5 | 7.16, dd (7.6, 6.8) | 124.4 |
| 7″ | 7.12, overlapped | 105.0 | 7.14, d (6.8) | 106.3 |
| 7″a | 137.9 | 140.0 | ||
| 2-SCH3 | 2.59, s | 14.0 | ||
| 2-SOCH3 | 2.98, s | 42.1 | ||
| 6′-OCH3 | 3.81, s | 55.2 | 3.89, s | 56.1 |
| 4″-OCH3 | 3.52, s | 54.9 | 3.55, s | 55.7 |
| 8 | |||||
|---|---|---|---|---|---|
| Position | δH, Multiplicity (J in Hz) a | δC | Position | δH, Multiplicity (J in Hz) a | δC |
| 1 | 157.1 | 9 | 158.8 | ||
| 2 | 6.80, d (8.0) | 101.5 | 10 | 7.02, d (2.4) | 97.5 |
| 3 | 7.40, t (8.0) | 128.2 | 10a | 144.6 | |
| 4 | 7.24, d (8.0) | 105.1 | NH-11 | 10.39, br s | |
| 4a | 143.4 | 11a | 135.8 | ||
| NH-5 | 10.86, br s | 12 | 8.53, s | 110.2 | |
| 5a | 138.0 | 12a | 122.0 | ||
| 6 | 82.5 | 12b | 113.0 | ||
| 6a | 123.0 | 1-OCH3 | 4.12, s | 56.0 | |
| 6b | 115.4 | 6-CN | 118.3 | ||
| 7 | 8.33, d (8.8) | 122.6 | 9-OH | 8.70, br s | |
| 8 | 6.86, dd (8.8, 2.4) | 110.0 | |||
| Compound | IC50 of NO Inhibition (μM) a | Cytotoxicity (μM) c |
|---|---|---|
| 1 | 186.4 ± 22.5 | >200 |
| 2 | 186.8 ± 23.1 | >200 |
| 3 | n.d. b | toxicity at 100 |
| 4 | >200 (231.2 ± 20.1 ***) | >200 |
| 5 | 92.2 ± 8.9 ** | >200 |
| 6 | n.d. b | toxicity at 100 |
| 7 | 31.1 ± 1.8 **** | toxicity at 100 |
| 8 | 56.7 ± 3.8 **** | >200 |
| indomethacin | 150.0 ± 16.0 | >200 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nukulkit, S.; Jantimaporn, A.; Poldorn, P.; Khongkow, M.; Rungrotmongkol, T.; Chang, H.-S.; Suttisri, R.; Chansriniyom, C. Eight Indole Alkaloids from the Roots of Maerua siamensis and Their Nitric Oxide Inhibitory Effects. Molecules 2022, 27, 7558. https://doi.org/10.3390/molecules27217558
Nukulkit S, Jantimaporn A, Poldorn P, Khongkow M, Rungrotmongkol T, Chang H-S, Suttisri R, Chansriniyom C. Eight Indole Alkaloids from the Roots of Maerua siamensis and Their Nitric Oxide Inhibitory Effects. Molecules. 2022; 27(21):7558. https://doi.org/10.3390/molecules27217558
Chicago/Turabian StyleNukulkit, Sasiwimon, Angkana Jantimaporn, Preeyaporn Poldorn, Mattaka Khongkow, Thanyada Rungrotmongkol, Hsun-Shuo Chang, Rutt Suttisri, and Chaisak Chansriniyom. 2022. "Eight Indole Alkaloids from the Roots of Maerua siamensis and Their Nitric Oxide Inhibitory Effects" Molecules 27, no. 21: 7558. https://doi.org/10.3390/molecules27217558