
Reinvestigation of Reactions of HgPh2 with Eu and Yb Metal and the Synthesis of a Europium(II) Bis(tetraphenylborate) †

Abstract
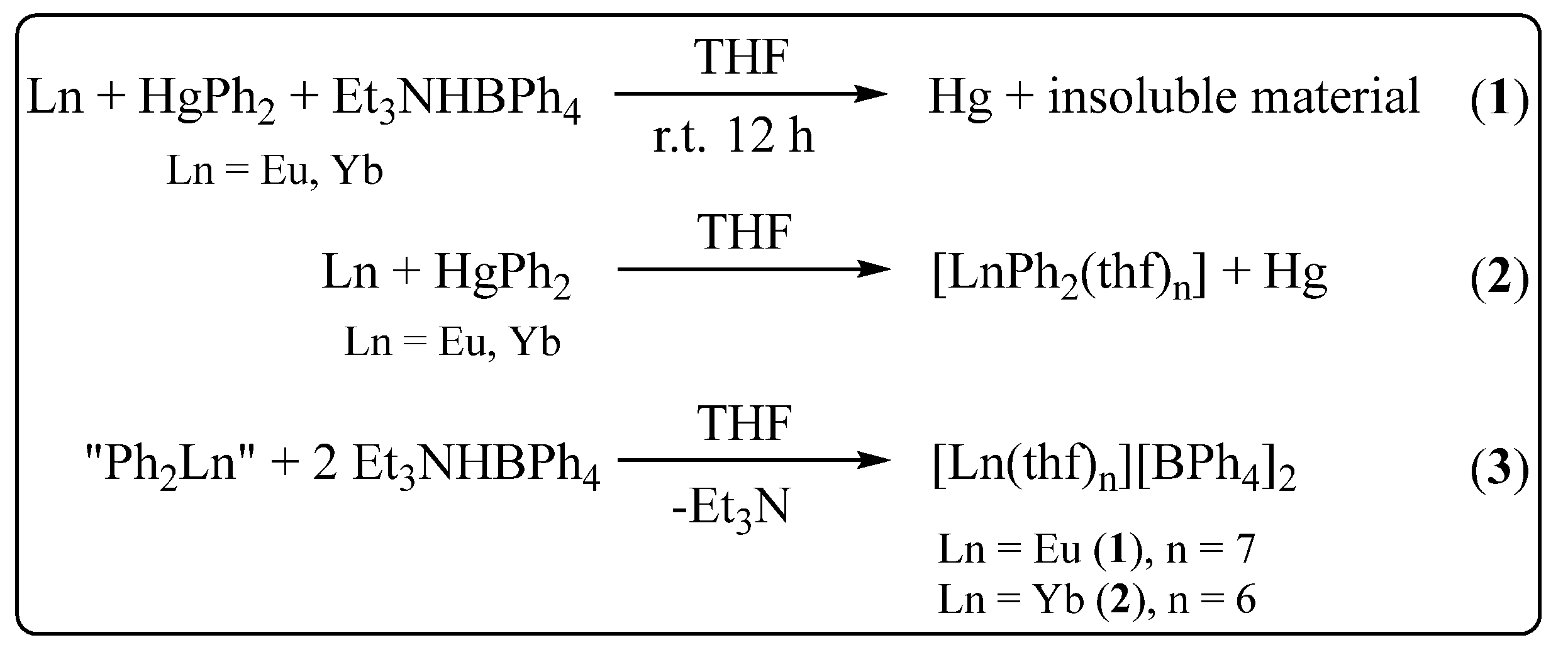
:1. Introduction
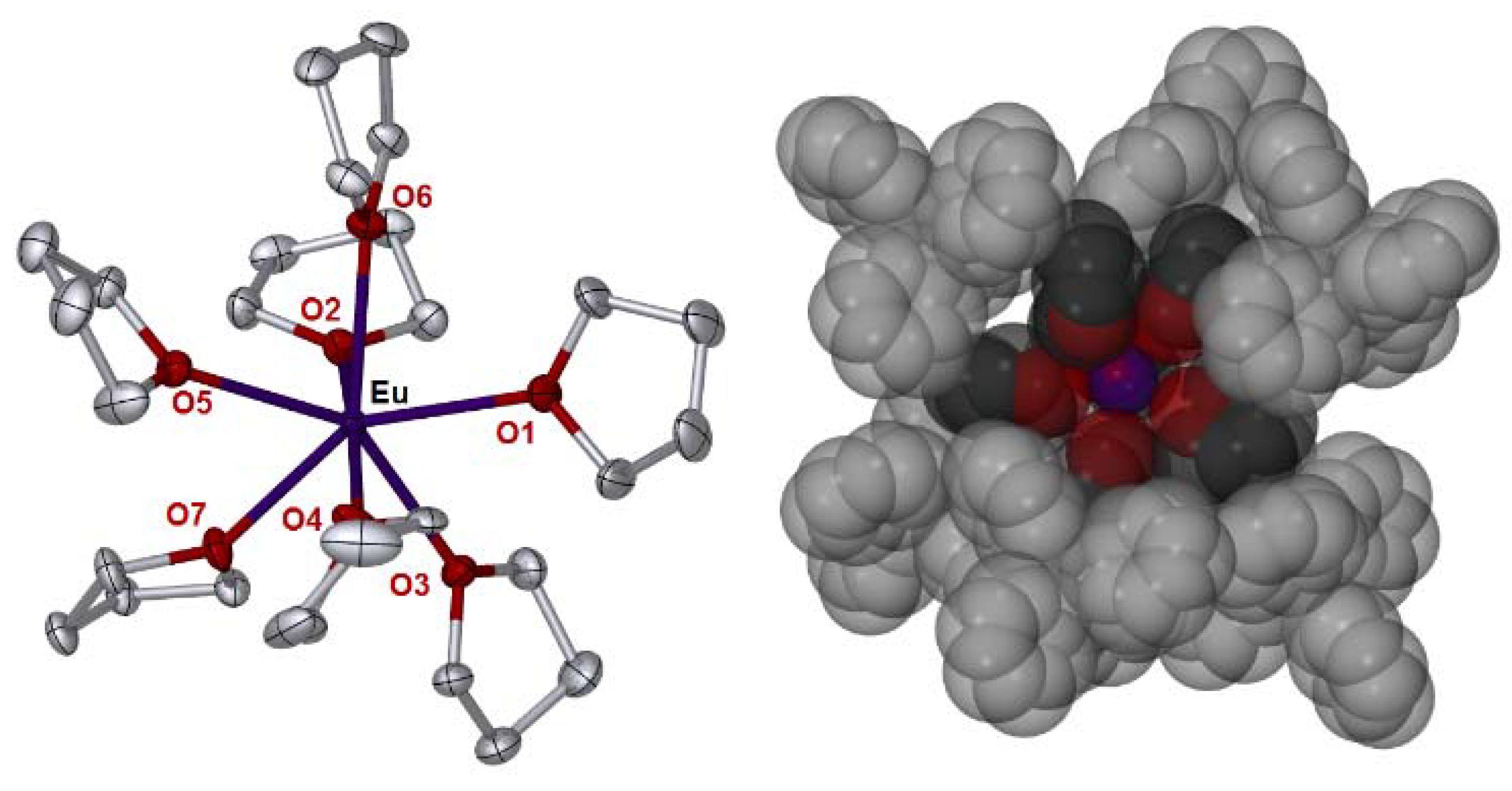
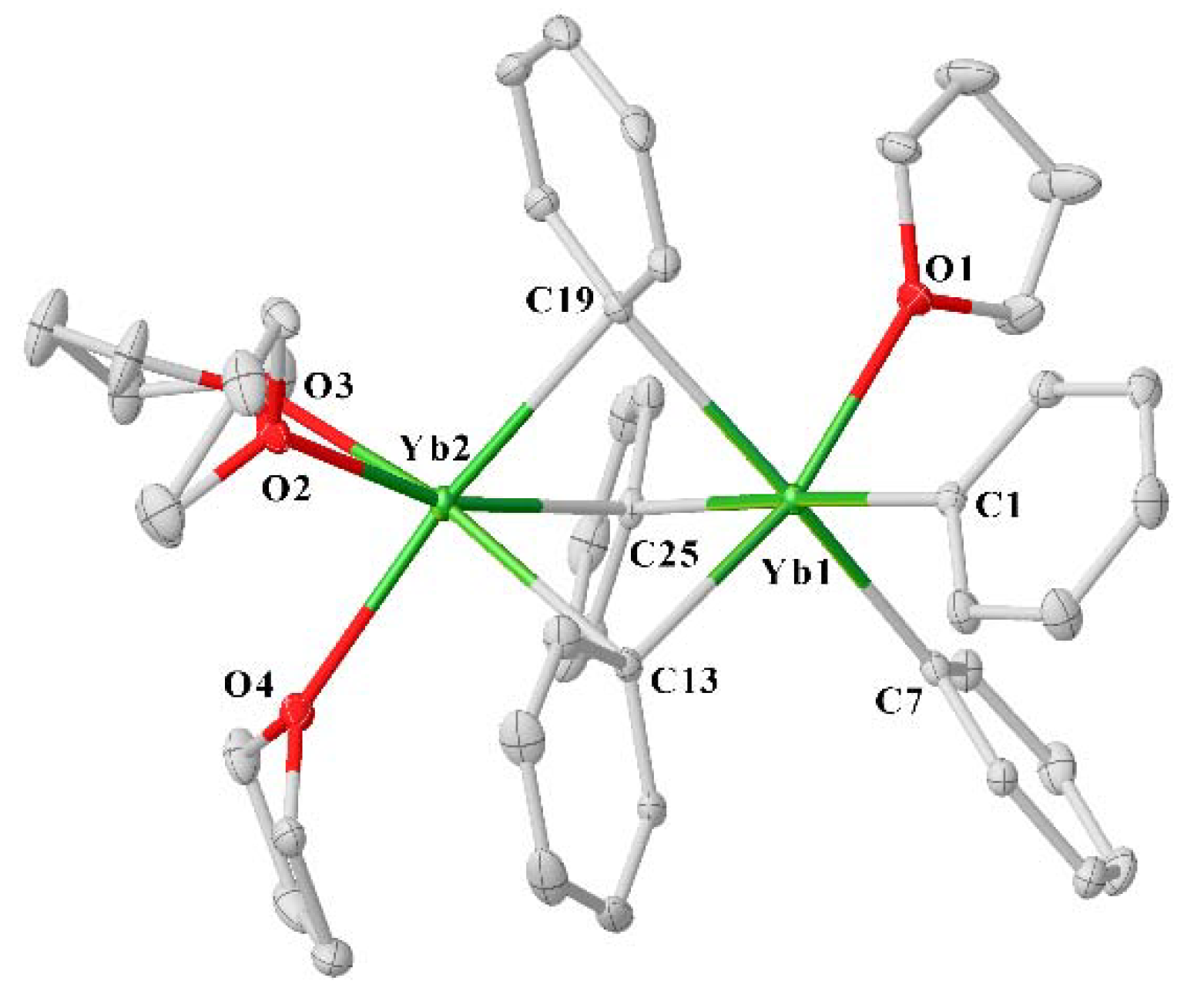
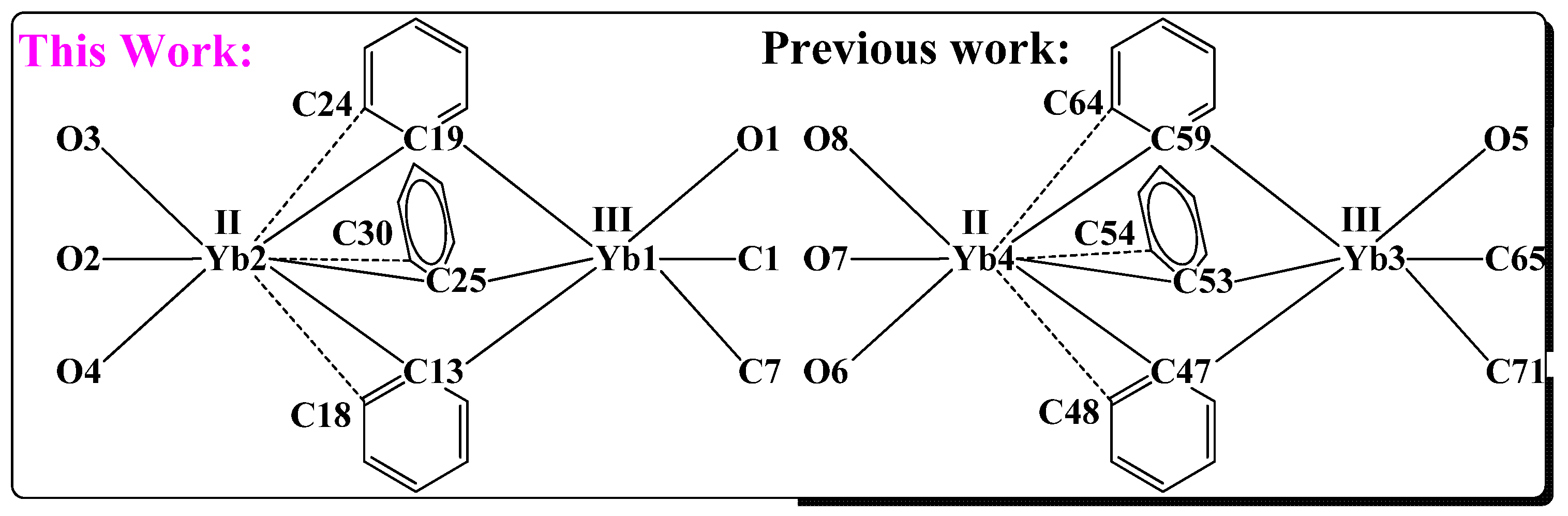
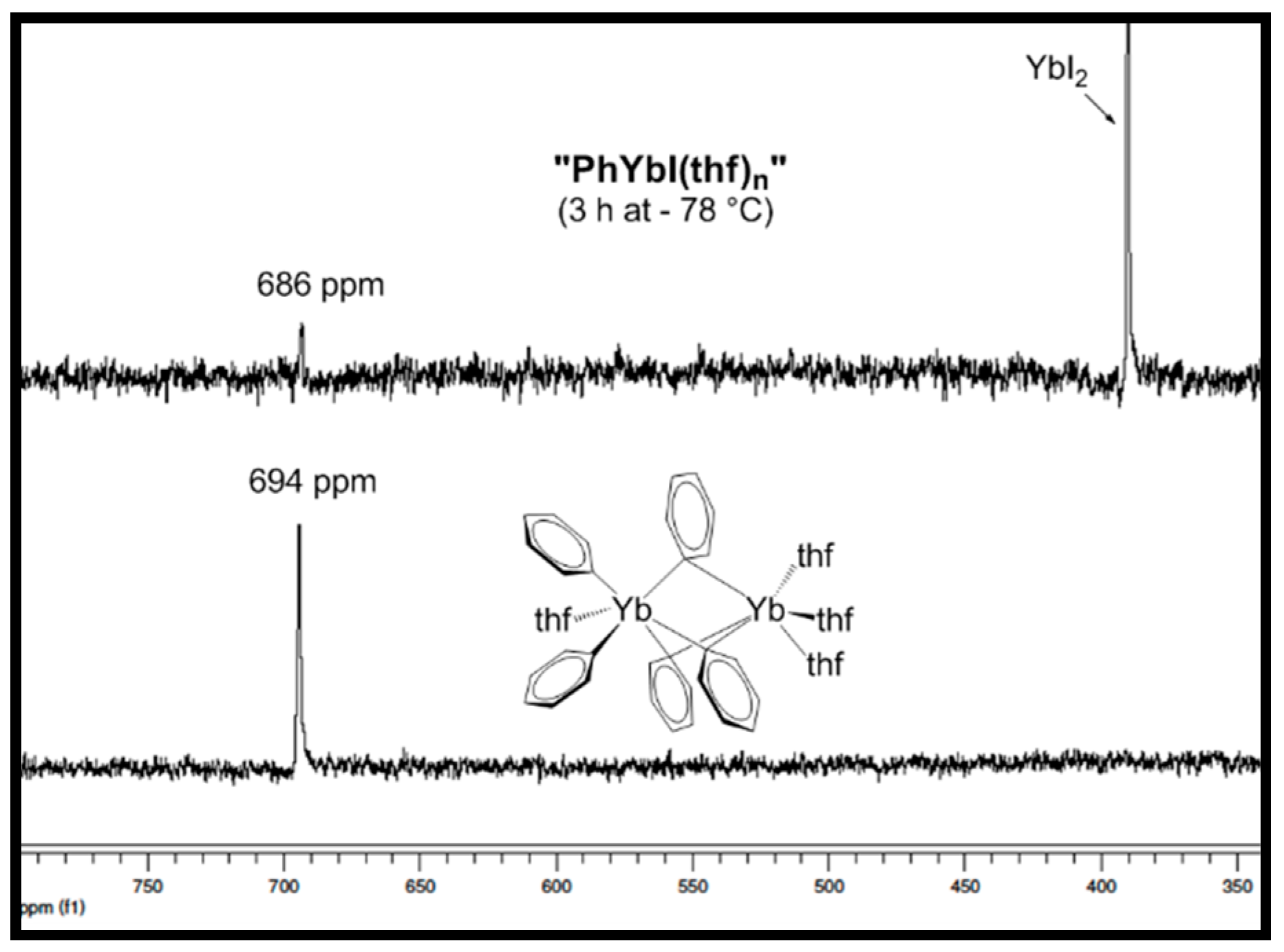
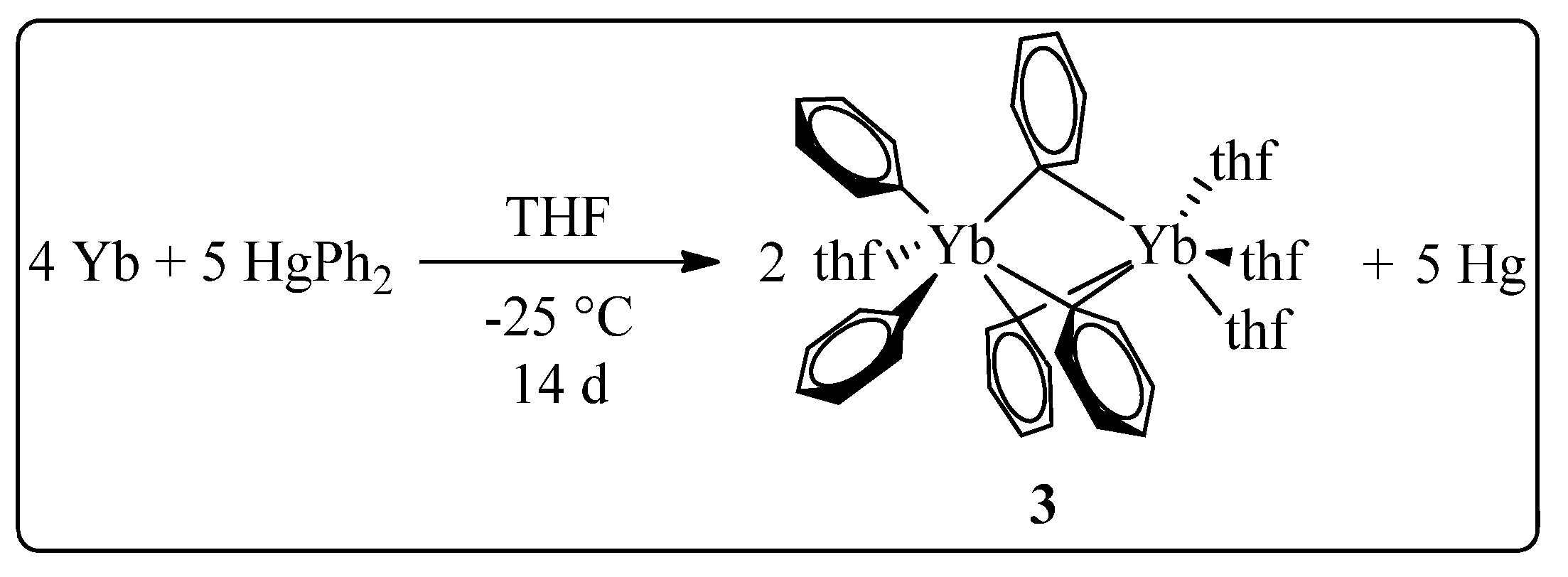
2. Results
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis of [Eu(thf)7][BPh4]2⋅thf (1)
3.3. Synthesis of [Yb(thf)6][BPh4]2 (2)
3.4. Synthesis of [Ph5Yb2(thf)4]⋅2thf (3)
3.5. X-ray Crystallography
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ortu, F. Rare Earth Starting Materials and Methodologies for Synthetic Chemistry. Chem. Rev. 2022, 122, 6040–6116. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Huo, R.; Tan, Y.Q.; Blair, V.; Deacon, G.B.; Junk, P.C. Syntheses of reactive rare earth complexes by redox transmetallation/protolysis reactions–A simple and convenient method. Coord. Chem. Rev. 2020, 415, 213232. [Google Scholar] [CrossRef]
- Zimmermann, M.; Anwander, R. Homoleptic rare-earth metal complexes containing Ln− C σ-bonds. Chem. Rev. 2010, 110, 6194–6259. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.R.; Hayes, P.G. Synthesis and reactivity of dialkyl lutetium complexes supported by a novel bis (phosphinimine) carbazole pincer ligand. Organometallics 2009, 28, 6352–6361. [Google Scholar] [CrossRef]
- Petrov, A.R.; Rufanov, K.A.; Harms, K.; Sundermeyer, J. Re-investigation of ortho-metalated N, N-dialkylbenzylamine complexes of rare-earth metals. First structurally characterized arylates of neodymium and gadolinium Li [LnAr4]. J. Organomet. Chem. 2009, 694, 1212–1218. [Google Scholar] [CrossRef]
- Yan, K.; Upton, B.M.; Ellern, A.; Sadow, A.D. Lewis acid-mediated β-hydride abstraction reactions of divalent M(C(SiHMe2)3)2THF2 (M= Ca, Yb). J. Am. Chem. Soc. 2009, 131, 15110–15111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, S.; Meetsma, A.; Hessen, B. Highly efficient hydrosilylation of alkenes by organoyttrium catalysts with sterically demanding amidinate and guanidinate ligands. Organometallics 2008, 27, 3131–3135. [Google Scholar] [CrossRef] [Green Version]
- Lyubov, D.M.; Fukin, G.K.; Trifonov, A.A. N,N′-Diisopropyl-N′′-bis(trimethylsilyl)guanidinate Ligand as a Supporting Coordination Environment in Yttrium Chemistry. Synthesis, Structure, and Properties of Complexes [(Me3Si)2NC(Ni-Pr)2]YCl2(THF)2,[(Me3Si)2NC(Ni-Pr)2]Y(CH2SiMe3)2(THF)2, and [(Me3Si)2NC(Ni-Pr)2]Y[(μ-H)(μ-Et)2BEt]2(THF). Inorg. Chem. 2007, 46, 11450–11456. [Google Scholar]
- Bambirra, S.; Meetsma, A.; Hessen, B. Lanthanum tribenzyl complexes as convenient starting materials for organolanthanum chemistry. Organometallics 2006, 25, 3454–3462. [Google Scholar] [CrossRef] [Green Version]
- Buschmann, D.A.; Schumacher, L.; Anwander, R. Rare-earth-metal half-sandwich complexes incorporating methyl, methylidene, and hydrido ligands. Chem. Commun. 2022, 58, 9132–9135. [Google Scholar] [CrossRef]
- Molander, G.A.; Romero, J.A.C. Lanthanocene catalysts in selective organic synthesis. Chem. Rev. 2002, 102, 2161–2186. [Google Scholar] [PubMed]
- Gromada, J.; Carpentier, J.F.; Mortreux, A. Group 3 metal catalysts for ethylene and α-olefin polymerization. Coord. Chem. Rev. 2004, 248, 397–410. [Google Scholar] [CrossRef]
- Hou, Z.; Wakatsuki, Y. Recent developments in organolanthanide polymerization catalysts. Coord. Chem. Rev. 2002, 231, 1–22. [Google Scholar] [CrossRef]
- Trambitas, A.G.; Panda, T.K.; Jenter, J.; Roesky, P.W.; Daniliuc, C.; Hrib, C.G.; Jones, P.G.; Tamm, M. Rare-earth metal alkyl, amido, and cyclopentadienyl complexes supported by imidazolin-2-iminato ligands: Synthesis, structural characterization, and catalytic application. Inorg. Chem. 2010, 49, 2435–2446. [Google Scholar] [CrossRef]
- Wooles, A.J.; Mills, D.P.; Lewis, W.; Blake, A.J.; Liddle, S.T. Lanthanide tri-benzyl complexes: Structural variations and useful precursors to phosphorus-stabilised lanthanide carbenes. Dalton Trans. 2010, 39, 500–510. [Google Scholar] [CrossRef]
- Ge, S.; Meetsma, A.; Hessen, B. Neutral and Cationic Rare Earth Metal Alkyl and Benzyl Compounds with the 1, 4, 6-Trimethyl-6-pyrrolidin-1-yl-1,4-diazepane Ligand and Their Performance in the Catalytic Hydroamination/Cyclization of Aminoalkenes. Organometallics 2008, 27, 5339–5346. [Google Scholar] [CrossRef] [Green Version]
- Zeimentz, P.M.; Okuda, J. Cationic aryl complexes of the rare-earth metals. Organometallics 2007, 26, 6388–6396. [Google Scholar] [CrossRef]
- Arndt, S.; Okuda, J. Cationic Alkyl Complexes of the Rare-Earth Metals: Synthesis, Structure, and Reactivity. Adv. Synth. Catal. 2005, 347, 339–354. [Google Scholar]
- Zeimentz, P.M.; Arndt, S.; Elvidge, B.R.; Okuda, J. Cationic organometallic complexes of scandium, yttrium, and the lanthanoids. Chem. Rev. 2006, 106, 2404–2433. [Google Scholar] [CrossRef]
- Robert, D.; Spaniol, T.P.; Okuda, J. Neutral and Monocationic Half-Sandwich Methyl Rare-Earth Metal Complexes: Synthesis, Structure, and 1,3-Butadiene Polymerization Catalysis. Eur. J. Inorg. Chem. 2008, 2008, 2801–2809. [Google Scholar]
- Nishiura, M.; Mashiko, T.; Hou, Z. Synthesis and styrene polymerisation catalysis of η5-and η1-pyrrolyl-ligated cationic rare earth metal aminobenzyl complexes. Chem. Commun. 2008, 2019–2021. [Google Scholar] [CrossRef] [PubMed]
- Döring, C.; Kretschmer, W.P.; Bauer, T.; Kempe, R. Scandium Aminopyridinates: Synthesis, Structure and Isoprene Polymerization. Eur. J. Inorg. Chem. 2009, 2009, 4255–4264. [Google Scholar] [CrossRef]
- Zimmermann, M.; Toörnroos, K.W.; Waymouth, R.M.; Anwander, R. Structure-Reactivity Relationships of Amido-Pyridine-Supported Rare-Earth-Metal Alkyl Complexes. Organometallics 2008, 27, 4310–4317. [Google Scholar] [CrossRef]
- Li, X.; Nishiura, M.; Mori, K.; Mashiko, T.; Hou, Z. Cationic scandium aminobenzyl complexes. Synthesis, structure and unprecedented catalysis of copolymerization of 1-hexene and dicyclopentadiene. Chem. Commun. 2007, 4137–4139. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Seibel, C.A.; Ziller, J.W. Unsolvated Lanthanide Metallocene Cations [(C5Me5)2Ln][BPh4]: Multiple Syntheses, Structural Characterization, and Reactivity Including the Formation of (C5Me5)3Nd1. J. Am. Chem. Soc. 1998, 120, 6745–6752. [Google Scholar] [CrossRef]
- Evans, W.J.; Champagne, T.M.; Ziller, J.W. Synthesis and reactivity of mono(pentamethylcyclopentadienyl) tetraphenylborate lanthanide complexes of ytterbium and samarium: Tris (ring) precursors to (C5Me5) Ln moieties. Organometallics 2007, 26, 1204–1211. [Google Scholar] [CrossRef]
- Evans, W.J.; Walensky, J.R.; Champagne, T.M.; Ziller, J.W.; DiPasquale, A.G.; Rheingold, A.L. Displacement, reduction, and ligand redistribution reactivity of the cationic mono-C5Me5 Ln2+ complexes (C5Me5) Ln (BPh4)(Ln= Sm, Yb). J. Organomet. Chem. 2009, 694, 1238–1243. [Google Scholar] [CrossRef]
- Wiecko, M.; Roesky, P.W. A Cationic Bis (phosphiniminomethanide) Europium (II) Complex. Organometallics 2009, 28, 1266–1269. [Google Scholar] [CrossRef]
- Deacon, G.B.; Evans, D.J.; Forsyth, C.M.; Junk, P.C. Lanthanoid (II) tetraphenylborate complexes: From discrete ions to pseudo metallocenes. Coord. Chem. Rev. 2007, 251, 1699–1706. [Google Scholar] [CrossRef]
- Deacon, G.B.; Forsyth, C.M. Synthesis and structures of the first cationic perfluoroaryllanthanoid (II) complexes. Chem. Eur. J. 2004, 10, 1798–1804. [Google Scholar] [CrossRef]
- Evans, W.J.; Johnston, M.A.; Greci, M.A.; Gummersheimer, T.S.; Ziller, J.W. Divalent lanthanide complexes free of coordinating anions: Facile synthesis of fully solvated dicationic [LnLx]2+ compounds. Polyhedron 2003, 22, 119–126. [Google Scholar]
- Starostina, T.A.; Shifrina, R.R.; Rybakova, L.F.; Petrov, E.S. Diphenyl ytterbium. Zh. Obshch. Khim. 1987, 57, 2402. [Google Scholar]
- Rybakova, L.F.; Tsygankova, S.V.; Rojtershtejn, D.M.; Petrov, E.S. Metallation of 1,3-diphenyl-2-benzylpropene by diphenylytterbium. Russ. J. Gen. Chem. 2004, 74, 141. [Google Scholar]
- Shannon, R.D. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta crystallographica section A: Crystal physics, diffraction, theoretical and general crystallography. Acta. Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Cole, M.L.; Deacon, G.B.; Forsyth, C.M.; Junk, P.C.; Konstas, K.; Wang, J. Steric modulation of coordination number and reactivity in the synthesis of lanthanoid (III) formamidinates. Chem. Eur. J. 2007, 13, 8092–8110. [Google Scholar] [PubMed]
- Evans, W.J.; Allen, N.T.; Ziller, J.W. Facile dinitrogen reduction via organometallic Tm (II) chemistry. J. Am. Chem. Soc. 2001, 123, 7927–7928. [Google Scholar] [CrossRef]
- Guo, Z.; Blair, V.; Deacon, G.B.; Junk, P.C. Can Bismuth Replace Mercury in Redox Transmetallation/Protolysis Syntheses from Free Lanthanoid Metals? Chem. Eur. J. 2018, 24, 17464–17474. [Google Scholar] [CrossRef]
- Guo, Z.; Blair, V.; Deacon, G.B.; Junk, P.C. Widely contrasting outcomes from the use of tris (pentafluorophenyl) bismuth or pentafluorophenylsilver as oxidants in the reactions of lanthanoid metals with N, N′-diarylformamidines. Dalton Trans. 2020, 49, 13588–13600. [Google Scholar] [CrossRef]
- Bochkarev, M.N.; Khramenkov, V.V.; Rad’kov, Y.F.; Zakharov, L.N.; Struchkov, Y.T. Synthesis and characterization of pentaphenyldiytterbium Ph2Yb(THF)(μ-Ph)3Yb(THF)3. J. Organomet. Chem. 1992, 429, 27–39. [Google Scholar]
- Wiecko, M.; Deacon, G.B.; Junk, P.C. Organolanthanoid-halide synthons−a new general route to monofunctionalized lanthanoid (ii) compounds? Chem. Commun. 2010, 46, 5076–5078. [Google Scholar]
- Ali, S.H.; Deacon, G.B.; Junk, P.C.; Hamidi, S.; Wiecko, M.; Wang, J. Lanthanoid Pseudo−Grignard Reagents: A Major Untapped Resource. Chem. Eur. J. 2018, 24, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Deacon, G.B.; Forsyth, C.M. A half-sandwich perfluoro-organoytterbium(II) complex from a simple redox transmetallation/ligand exchange synthesis. Organometallics 2003, 22, 1349–1352. [Google Scholar] [CrossRef]
- Bochkarev, L.N.; Zheleznova, T.A.; Safronova, A.V.; Drozdov, M.S.; Zhil′tsov, S.F.; Zakharov, L.N.; Fukin, G.K.; Khorshev, S.Y. Synthesis and crystal structure of triphenyl[tris(tetrahydrofuran)]ytterbium, Ph3Yb(THF)3. Russ. Chem. Bull. 1998, 47, 163–166. [Google Scholar] [CrossRef]
- Deacon, G.B.; Shen, Q. Complexes of lanthanoids with neutral π donor ligands. J. Organomet. Chem. 1996, 511, 1–17. [Google Scholar] [CrossRef]
- Cloke, F.G.N. Zero oxidation state compounds of scandium, yttrium, and the lanthanides. Chem. Soc. Rev. 1993, 22, 17–24. [Google Scholar] [CrossRef]
- Liang, H.; Shen, Q.; Jin, S.; Lin, Y. Synthesis and X-ray crystal structure of [Eu(η6-C6Me6)(AlCl4)2]4; the first cyclotetrameric lanthanide(II) complex with a neutral π-ligand. J. Chem. Soc. Chem. Commun. 1992, 480–481. [Google Scholar] [CrossRef]
- Deacon, G.B.; Forsyth, C.M.; Junk, P.C.; Skelton, B.W.; White, A.H. The striking influence of intramolecular lanthaoid π-arene interactions on the structural architecture of the homoleptic aryloxolanthanoid(II) complexes, [Eu2(Odpp)(μ-Odpp)3] and [Yb2(Odpp)2(μ-Odpp)2], and the Yb(II)–Yb(III) trimetallic [Yb2(μ-Odpp)3][Yb(Odpp)4]- (Odpp = 2,6-diphenylphenolato). Chem. Eur. J. 1999, 5, 1452–1459. [Google Scholar]
- Deacon, G.B.; Forsyth, C.M. Linkage isomerism and C-H activation in an ytterbium(II) tetraphenylborate. Chem. Commun. 2002, 2522–2523. [Google Scholar]
- Deacon, G.B.; Junk, P.C.; Moxey, G.J.; Ruhlandt-Senge, K.; Prix, C.S.; Zuniga, M.F. Charge-separated and molecular heterobimetallic rare earth-rare earth and alkaline-earth-rare earth aryloxo complexes featuring intramolecular metal -π-arene interactions. Chem. Eur. J. 2009, 15, 5503–5519. [Google Scholar] [CrossRef]
- Bondi, A. van der Waals volumes and radii. J. Phys. Chem. 1964, 68, 441–451. [Google Scholar] [CrossRef]
- Wells, A.F. Structural Inorganic Chemistry, 5th ed; Oxford University Press: Oxford, UK, 1984; p. 1288. [Google Scholar]
- Heckmann, G.; Niemeyer, M. Synthesis and first structural characterization of lanthanide (II) aryls: Observation of a Schlenk equilibrium in europium(II) and ytterbium(II) chemistry. J. Am. Chem. Soc. 2000, 122, 4227–4228. [Google Scholar] [CrossRef]
- Avent, A.G.; Edelman, M.A.; Lappert, M.F.; Lawless, G.A. The first high resolution direct NMR observation of an f-block element. J. Am. Chem. Soc. 1989, 111, 3423–3425. [Google Scholar] [CrossRef]
- Deacon, G.B.; Fallon, G.D.; Forsyth, C.M.; Schumann, H.; Weimann, R. Organoamido- and Aryloxo-lanthanoids. 15. Syntheses of low coordination number divalent lanthanoid organoamide complexes and the X-ray crystal structures of Bis[(N-2,6-di-iso-propylphenyl)(N-trimethylsilyl)amido]bis(tetrahydrofuran)-samarium(II) and -ytterbium(II). Chem. Ber. 1997, 130, 409–415. [Google Scholar]
- Sheldrick, G.M. SADABS, Software for Empirical Absorption Corrections; Standard Software Reference; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| This Work | Ref [39] * | ||
|---|---|---|---|
| Bond | Length (Å) | Bond | Length (Å) |
| Yb1-C1 | 2.446 (8) | Yb3-C65 | 2.39 (4) |
| Yb1-C7 | 2.445 (7) | Yb3-C71 | 2.46 (3) |
| Yb1-O1 | 2.400 (6) | Yb3-O5 | 2.30 (2) |
| Yb1-C13 | 2.489 (9) | Yb3-C59 | 2.48 (3) |
| Yb1-C19 | 2.573 (8) | Yb3-C53 | 2.54 (3) |
| Yb1-C25 | 2.583 (8) | Yb3-C47 | 2.51 (4) |
| Yb2-C13 | 2.665 (8) | Yb4-C59 | 2.68 (3) |
| Yb2-C19 | 2.641 (10) | Yb4-C53 | 2.55 (3) |
| Yb2-C25 | 2.602 (8) | Yb4-C47 | 2.75 (4) |
| Yb1-O4 | 2.452 (6) | Yb4-O6 | 2.44 (2) |
| Yb2-O2 | 2.434 (6) | Yb4-O7 | 2.38 (3) |
| Yb2-O3 | 2.462 (6) | Yb4-O8 | 2.50 (3) |
| Yb2…C18 | 3.034 (4) | Yb4…C48 | 3.08 (4) |
| Yb2…C24 | 2.995 (4) | Yb4…C64 | 2.93 (3) |
| Yb2…C30 | 3.130 (4) | Yb4…C54 | 3.25 (4) |
| Yb1-C14 a | 3.350 (4) | Yb3-C52 a | 3.45 (4) |
| Yb1-C20 a | 3.244 (4) | Yb3-C60 a | 3.16 (4) |
| Yb1-C26 a | 3.412 (4) | Yb3-C58 a | 3.39 (5) |
| Yb2…H24 | 2.785 | ||
| Yb2…H18 | 2.921 | ||
| Yb2…H30 | 3.060 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiecko, M.; Guo, Z.; Deacon, G.B.; Junk, P.C. Reinvestigation of Reactions of HgPh2 with Eu and Yb Metal and the Synthesis of a Europium(II) Bis(tetraphenylborate). Molecules 2022, 27, 7547. https://doi.org/10.3390/molecules27217547
Wiecko M, Guo Z, Deacon GB, Junk PC. Reinvestigation of Reactions of HgPh2 with Eu and Yb Metal and the Synthesis of a Europium(II) Bis(tetraphenylborate). Molecules. 2022; 27(21):7547. https://doi.org/10.3390/molecules27217547
Chicago/Turabian StyleWiecko, Michal, Zhifang Guo, Glen B. Deacon, and Peter C. Junk. 2022. "Reinvestigation of Reactions of HgPh2 with Eu and Yb Metal and the Synthesis of a Europium(II) Bis(tetraphenylborate)" Molecules 27, no. 21: 7547. https://doi.org/10.3390/molecules27217547