
New Trends in the Quality Control of Enantiomeric Drugs: Quality by Design-Compliant Development of Chiral Capillary Electrophoresis Methods

 , , , , and
, , , , and
Abstract
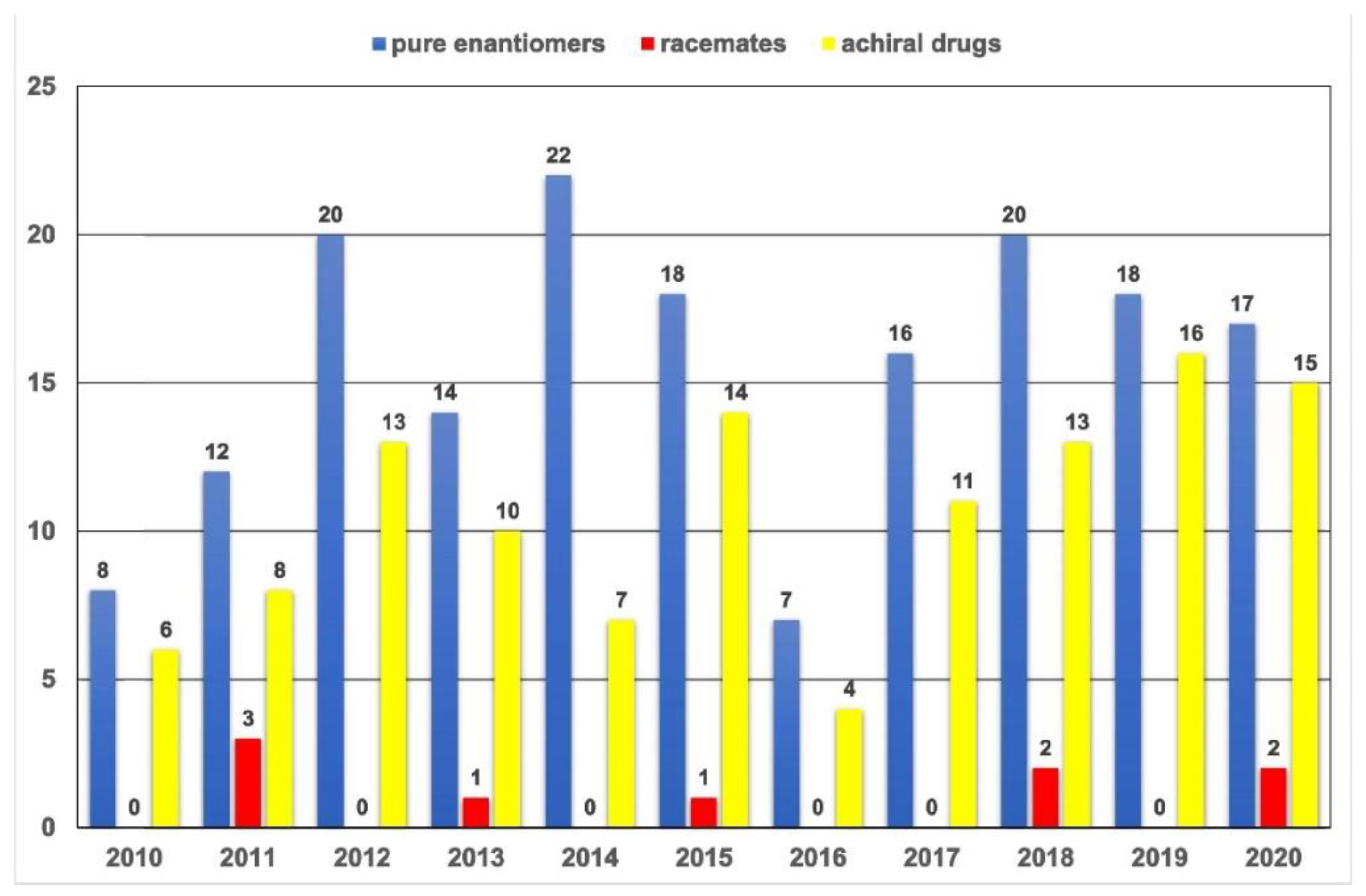
:1. Introduction
2. Recent Advances in the Development of CE Analytical Procedures for Enantiomeric Purity Control of Pharmaceuticals
2.1. Regulatory Requirements for Quality Purposes
2.2. ICH Q14 and ICH Q2(R2) Guidelines
2.2.1. ICH Q14 Enhanced Approach
2.2.2. ICH Q2(R2) Outlook
- -
- The term “Selectivity” is placed beside “Specificity”, covering the common possibility of uncomplete discrimination of the analytes, but at the same time giving value to the minimization of interference, even if the method cannot be proved as “specific”;
- -
- The Lower Range Limits, Detection Limit (DL) and Quantitation Limit (QL), are now included into the PC “Working Range”. In the case of impurity testing, when building the curve for assessing the relationship between analyte concentration and response, the lower concentration value of the Working Range should correspond to the QL;
- -
- As concerns Accuracy and Precision, there is the possibility of considering their total impact instead of evaluating them separately. The concept of Total Analytical Error has been mentioned, together with the AQbD approach, as a factor necessary for a quality scientific paper in drug analysis [46,49]; as a matter of fact, the uncertainty of the RR is related to Total Error;
- -
- For assessing Intermediate Precision, the variations tested (days, environmental conditions, analysts, equipment) should be based on AP understanding from development and risk assessment.
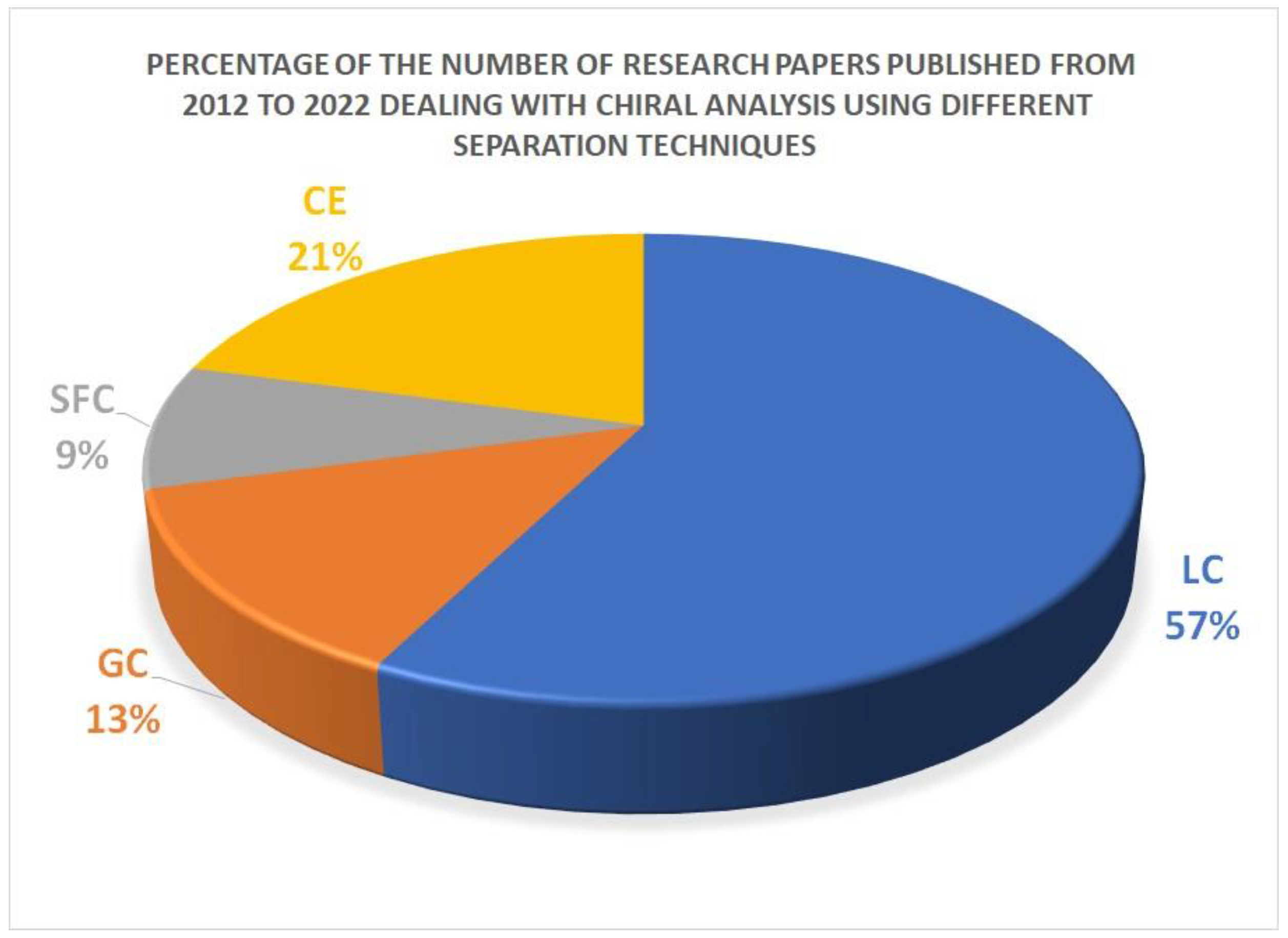
2.3. Capillary Electrophoresis for Enantiomeric Purity of Drugs
2.3.1. Chiral Selectors in EKC and Separation Mechanisms
2.3.2. Selected Examples of Chiral EKC of Drugs
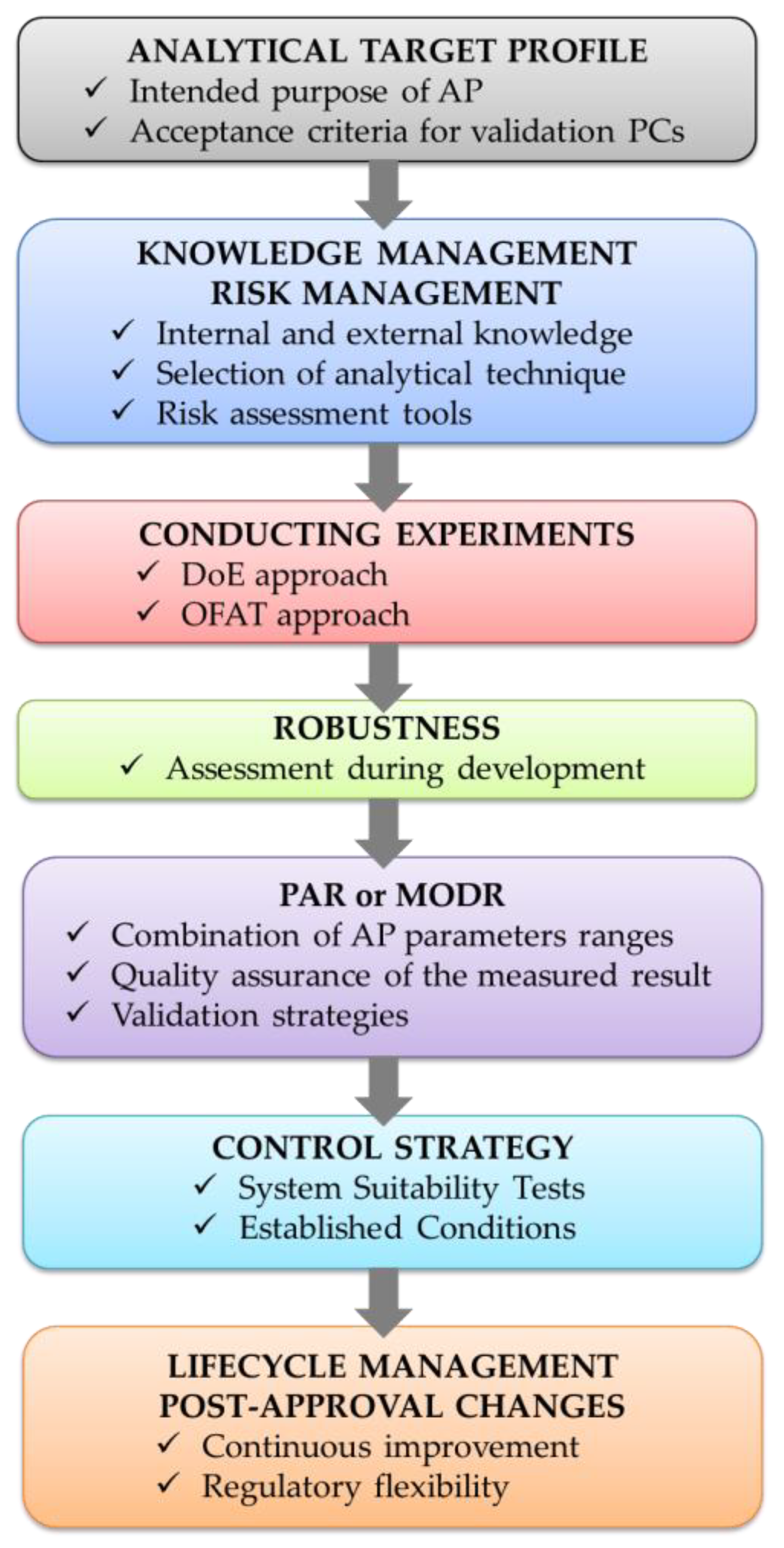
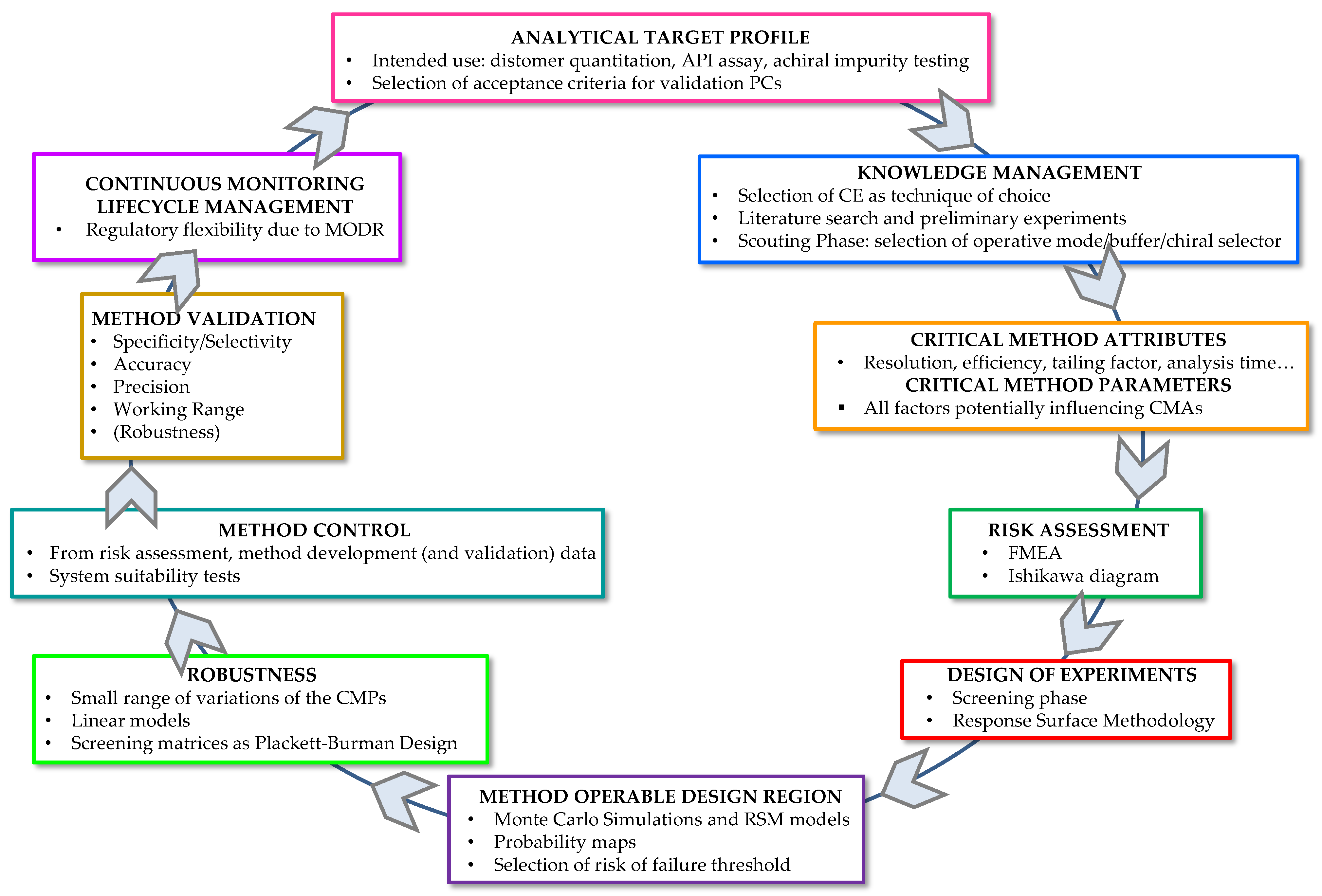
2.4. AQbD Framework in the Development of CE Analytical Procedures
2.4.1. Analytical Target Profile
2.4.2. Knowledge Management
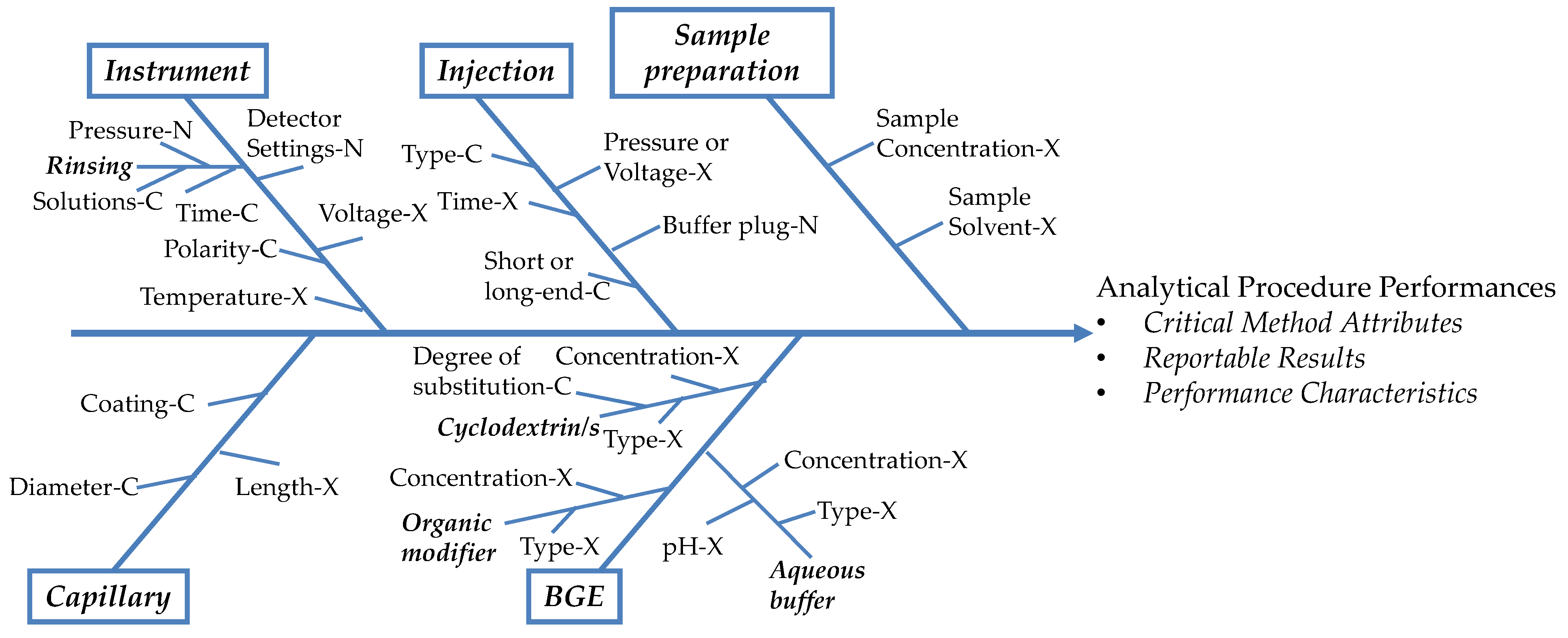
2.4.3. Critical Method Attributes and Critical Method Parameters
2.4.4. Risk Assessment
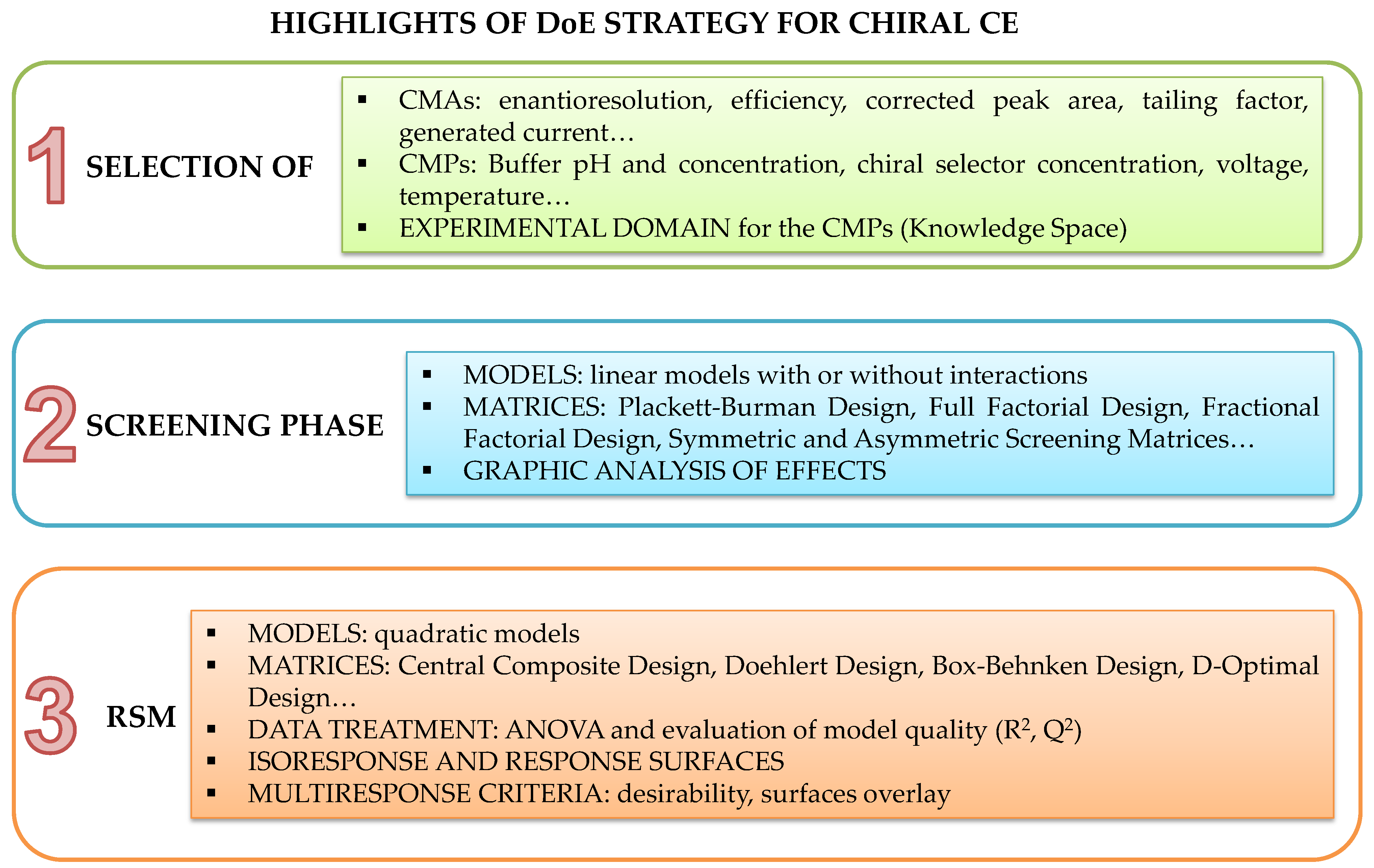
2.4.5. Design of Experiments
2.4.6. Method Operable Design Region
2.4.7. Robustness
2.4.8. Method Control
2.4.9. Method Validation
2.4.10. Continuous Monitoring—Lifecycle Management
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
Abbreviations
References
- Nguyen, L.A.; He, H.; Pham-Huy, C. Chiral drugs: An overview. Int. J. Biomed. Sci. 2006, 2, 85–100. [Google Scholar] [PubMed]
- Brooks, W.; Guida, W.; Daniel, K. The significance of chirality in drug design and development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.U. Chirality and its implications for the pharmaceutical industry. Rend. Lincei 2013, 24, 213–216. [Google Scholar] [CrossRef]
- De Camp, W.H. Chiral drugs: The FDA perspective on manufacturing and control. J. Pharm. Biomed. Anal. 1993, 11, 1167–1172. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs (accessed on 10 July 2022).
- Daniels, J.M.; Nestmann, E.R.; Kerr, A. Development of stereoisomers (chiral) drugs: A brief review of scientific and regulatory considerations. Drug Inf. J. 1997, 31, 639–646. [Google Scholar] [CrossRef]
- Calcaterra, A.; D’Acquarica, I. The market of chiral drugs: Chiral switches versus de novo enantiomerically pure compounds. J. Pharm. Biomed. Anal. 2018, 147, 323–340. [Google Scholar] [CrossRef]
- Agranat, I.; Caner, H.; Caldwell, J. Putting chirality to work: The strategy of chiral switches. Nat. Rev. Drug Discov. 2002, 1, 753–768. [Google Scholar] [CrossRef]
- Hancu, G.; Modroiu, A. Chiral Switch: Between therapeutical benefit and marketing strategy. Pharmaceuticals 2022, 15, 240. [Google Scholar] [CrossRef]
- Scriba, G.K. Chiral recognition in separation science–an update. J. Chromatogr. A 2016, 1467, 56–78. [Google Scholar] [CrossRef]
- Bernardo-Bermejo, S.; Sánchez-López, E.; Castro-Puyana, M.; Marina, M.L. Chiral capillary electrophoresis. Trends Anal. Chem. 2020, 124, 115807. [Google Scholar] [CrossRef]
- Fanali, S.; Chankvetadze, B. Some thoughts about enantioseparations in capillary electrophoresis. Electrophoresis 2019, 40, 2420–2437. [Google Scholar] [CrossRef]
- Yu, R.B.; Quirino, J.P. Chiral selectors in capillary electrophoresis: Trends during 2017–2018. Molecules 2019, 24, 1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saz, J.M.; Marina, M.L. Recent advances on the use of cyclodextrins in the chiral analysis of drugs by capillary electrophoresis. J. Chromat. A 2016, 1467, 79–94. [Google Scholar] [CrossRef] [PubMed]
- ICH Harmonised Tripartite Guideline. Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities) Q11; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2012. [Google Scholar]
- Mane, S. Racemic drug resolution: A comprehensive guide. Anal. Methods 2016, 8, 7567–7586. [Google Scholar] [CrossRef]
- Görög, S. Critical review of reports on impurity and degradation product profiling in the last decade. Trends Anal. Chem. 2018, 101, 2–16. [Google Scholar] [CrossRef]
- El Deeb, S.; Wätzig, H.; Abd El-Hady, D.; Sänger-van de Griend, C.; Scriba, G.K. Recent advances in capillary electrophoretic migration techniques for pharmaceutical analysis (2013–2015). Electrophoresis 2016, 37, 1591–1608. [Google Scholar] [CrossRef]
- Hancu, G.; Orlandini, S.; Papp, L.A.; Modroiu, A.; Gotti, R.; Furlanetto, S. Application of experimental design methodologies in the enantioseparation of pharmaceuticals by capillary electrophoresis: A review. Molecules 2021, 26, 4681. [Google Scholar] [CrossRef]
- Krait, S.; Konjaria, M.L.; Scriba, G.K. Advances of capillary electrophoresis enantioseparations in pharmaceutical analysis (2017–2020). Electrophoresis 2021, 42, 1709–1725. [Google Scholar] [CrossRef]
- ICH Harmonised Tripartite Guideline. Pharmaceutical Development Q8(R2); International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2009. [Google Scholar]
- Fukuda, I.M.; Francini Fidelis Pinto, C.; dos Santos Moreira, C.; Morais Saviano, A.; Rebello Lourenço, F. Design of experiments (DoE) applied to pharmaceutical and analytical Quality by Design (QbD). Braz. J. Pharm. Sci. 2018, 54, e01006. [Google Scholar] [CrossRef]
- ICH Harmonised Tripartite Guideline. Analytical Procedure Development Q14; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2022. [Google Scholar]
- ICH Harmonised Tripartite Guideline. Validation of Analytical Procedures Q2(R2); International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2022. [Google Scholar]
- Hubert, C.; Lebrun, P.; Houari, S.; Ziemons, E.; Rozet, E.; Hubert, P. Improvement of a stability-indicating method by Quality-by-Design versus Quality-by-Testing: A case of a learning process. J. Pharm. Biomed. Anal. 2014, 88, 401–409. [Google Scholar] [CrossRef]
- Pharmaceutical CGMPs for the 21st Century—A Risk-Based Approach Final Report; Department of Health and Human Services, U.S. Food and Drug Administration: Siler Spring, MD, USA, 2004.
- Guidance for Industry PAT—A Framework for Innovative Pharmaceutical Development, Manufacturing, and Quality Assurance; U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM), Office of Regulatory Affairs (ORA), Pharmaceutical CGMPs: Spring, MD, USA, 2004.
- ICH Harmonised Tripartite Guideline. Quality Risk Management Q9; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2005. [Google Scholar]
- ICH Harmonised Tripartite Guideline. Pharmaceutical Quality System Q10; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2008. [Google Scholar]
- Borman, P.; Truman, K.; Thompson, D.; Nethercote, P.; Chatfield, M. The application of Quality by Design to analytical methods. Pharm. Technol. 2007, 31, 142–152. [Google Scholar]
- Vogt, F.G.; Kord, A.S. Development of Quality-By-Design analytical methods. J. Pharm. Sci. 2011, 100, 797–812. [Google Scholar] [CrossRef] [PubMed]
- Monks, K.; Molnár, I.; Rieger, H.J.; Bogáti, B.; Szabó, E. Quality by Design: Multidimensional exploration of the design space in high performance liquid chromatography method development for better robustness before validation. J. Chromatogr. A 2012, 1232, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Tome, T.; Žigart, N.; Časar, Z.; Obreza, A. Development and optimization of liquid chromatography analytical methods by using AQbD principles: Overview and recent advances. Org. Process Res. Dev. 2019, 23, 1784–1802. [Google Scholar] [CrossRef] [Green Version]
- Rozet, E.; Lebrun, P.; Hubert, P.; Debrus, B.; Boulanger, B. Design Spaces for analytical methods. Trends Anal. Chem. 2013, 42, 157–167. [Google Scholar] [CrossRef]
- Orlandini, S.; Pinzauti, S.; Furlanetto, S. Application of quality by design to the development of analytical separation methods. Anal. Bioanal. Chem. 2013, 405, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Deidda, R.; Orlandini, S.; Hubert, P.; Hubert, C. Risk-based approach for method development in pharmaceutical quality control context: A critical review. J. Pharm. Biomed. Anal. 2018, 161, 110–121. [Google Scholar] [CrossRef]
- Breitkreitz, M.C. Analytical Quality by Design. Braz. J. Anal. Chem. 2021, 8, 1–5. [Google Scholar] [CrossRef]
- Hubert, C.; Houari, S.; Rozet, E.; Lebrun, P.; Hubert, P. Towards a full integration of optimization and validation phases: An analytical-quality-by-design approach. J. Chromatogr. A 2015, 1395, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Vedantika, D.; Bhushan, B.; Saudagar, R.B. Quality by Design approaches to Analytical Method Development. Res. J. Pharm. Technol. 2017, 10, 3188–3194. [Google Scholar]
- Jahangir, M.A.; Taleuzzaman, M.; Alam, M.J.; Soni, A.; Beg, S. Analytical quality by design for capillary electrophoresis. In Handbook of Analytical Quality by Design; Beg, S., Hasnain, M.S., Rahman, M., Almalki, W.H., Eds.; Elsevier Academic Press: Amsterdam, The Netherlands, 2021; pp. 115–132. [Google Scholar]
- Rozet, E.; Ziemons, E.; Marini, R.D.; Boulanger, B.; Hubert, P. Quality by design compliant analytical method validation. Anal. Chem. 2012, 84, 106–112. [Google Scholar] [CrossRef]
- Dispas, A.; Avohou, H.T.; Lebrun, P.; Hubert, P.; Hubert, C. ‘Quality by Design’ approach for the analysis of impurities in pharmaceutical drug products and drug substances. Trends Anal. Chem. 2018, 101, 24–33. [Google Scholar]
- Parr, M.K.; Schmidt, A.H. Life cycle management of analytical methods. J. Pharm. Biomed. Anal. 2018, 147, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Kovács, B.; Péterfi, O.; Kovács-Deák, B.; Székely-Szentmiklósi, I.; Fülöp, I.; Bába, L.I.; Boda, F. Quality-by-design in pharmaceutical development: From current perspectives to practical applications. Acta Pharm. 2021, 71, 497–526. [Google Scholar] [CrossRef]
- Borman, P.; Campa, C.; Delpierre, G.; Hook, E.; Jackson, P.; Kelley, W.; Protz, M.; Vandeputte, O. Selection of analytical technology and development of analytical procedures using the analytical target profile. Anal. Chem. 2022, 94, 559–570. [Google Scholar] [CrossRef]
- Verch, T.; Campa, C.; Chéry, C.C.; Frenkel, R.; Graul, T.; Jaya, N.; Nakhle, B.; Springall, J.; Starkey, J.; Wypych, J.; et al. Analytical Quality by Design, life cycle management, and method control. AAPS J. 2022, 24, 34. [Google Scholar] [CrossRef]
- Ermer, J.; Aguiar, D.; Boden, A.; Ding, B.; Obeng, D.; Rose, M.; Vokrot, J. Lifecycle management in pharmaceutical analysis: How to establish an efficient and relevant continued performance monitoring program. J. Pharm. Biomed. Anal. 2020, 181, 113051. [Google Scholar] [CrossRef]
- Volta e Sousa, L.; Gonçalves, R.; Menezes, J.C.; Ramos, A. Analytical method lifecycle management in pharmaceutical industry: A review. AAPS PharmSciTech 2021, 22, 128. [Google Scholar] [CrossRef]
- Dispas, A.; Hubert, C.; Hubert, P. Perspective: What constitutes a quality paper in drug analysis? Talanta Open 2021, 4, 100054. [Google Scholar] [CrossRef]
- Teasdale, A.; Popkin, M.; Ogilvie, R.; Borman, P.J.; Antonucci, V. Regulatory Highlights. Org. Process Res. Dev. 2018, 22, 1712–1715. [Google Scholar] [CrossRef]
- Goupy, J.L. Methods for Experimental Design, 1st ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Deming, S.N.; Morgan, S.L. Experimental Design: A Chemometric Approach, 2nd ed.; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1993. [Google Scholar]
- Massart, D.L.; Vandeginste, B.G.M.; Buydens, L.M.C.; De Jong, S.; Lewi, P.J.; Smeyers-Verbeke, J. Handbook of Chemometrics and Qualimetrics: Part A, 1st ed.; Elsevier Science B.V.: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Wikström, C.; Wold, S. Design of Experiments-Principles and Applications, 3rd ed.; Umetrics AB: Umeå, Sweden, 2008. [Google Scholar]
- Raman, N.V.S.; Mallu, U.R.; Bapatu, H.R. Analytical Quality by Design approach to test method development and validation in drug substance manufacturing. J. Chem. 2015, 2015, 435129. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Deshpande, M.; Zaherr, Z.; Shinde, D.B.; Arote, R. Quality by design approach: Regulatory need. Arab. J. Chem. 2017, 10, 3412–3425. [Google Scholar] [CrossRef] [Green Version]
- Teasdale, A.; Borman, P.; Mullen, A.K. Regulatory Highlights. Org. Process Res. Dev. 2022, 26, 1029–1037. [Google Scholar] [CrossRef]
- Chankvetadze, B. Contemporary theory of enantioseparations in capillary electrophoresis. J. Chromatogr. A 2018, 1567, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Gumustas, M.; Ozkan, S.A.; Chankvetadze, B. Analytical and preparative scale separation of enantiomers of chiral drugs by chromatography and related methods. Curr. Med. Chem. 2018, 25, 4152–4188. [Google Scholar] [CrossRef]
- Ilisz, I.; Aranyi, A.; Péter, A. Chiral derivatizations applied for the separation of unusual amino acid enantiomers by liquid chromatography and related techniques. J. Chromatogr. A 2013, 1296, 119–139. [Google Scholar] [CrossRef]
- Prior, A.; Moldovan, R.C.; Crommen, J.; Servais, A.C.; Fillet, M.; de Jong, G.J.; Somsen, G.W. Enantioselective capillary electrophoresis-mass spectrometry of amino acids in cerebrospinal fluid using a chiral derivatizing agent and volatile surfactant. Anal. Chim. Acta 2016, 940, 150–158. [Google Scholar] [CrossRef]
- Moldovan, R.C.; Bodoki, E.; Kacsó, T.; Servais, A.C.; Crommen, J.; Oprean, R.; Fillet, M. A micellar electrokinetic chromatography-mass spectrometry approach using in-capillary diastereomeric derivatization for fully automatized chiral analysis of amino acids. J. Chromatogr. A 2016, 1467, 400–408. [Google Scholar] [CrossRef]
- Moldovan, R.C.; Bodoki, E.; Servais, A.C.; Crommen, J.; Oprean, R.; Fillet, M. (+) or (-)-1-(9-fluorenyl)ethyl chloroformate as chiral derivatizing agent: A review. J. Chromatogr. A 2017, 1513, 1–17. [Google Scholar] [CrossRef]
- D’Orazio, G.; Fanali, C.; Asensio-Ramos, M.; Fanali, S. Chiral separations in food analysis. Trends Anal. Chem. 2017, 96, 151–171. [Google Scholar] [CrossRef]
- Caslavska, J.; Thormann, W. Bioanalysis of drugs and their metabolites by chiral electromigration techniques (2010–2020). Electrophoresis 2021, 42, 1744–1760. [Google Scholar] [CrossRef]
- Chankvetadze, B. Application of enantioselective separation techniques to bioanalysis of chiral drugs and their metabolites. Trends Anal. Chem. 2021, 143, 116332. [Google Scholar] [CrossRef]
- Kartsova, L.A.; Moskvichev, D.O. In-capillary chiral derivatization of amino acids. J. Anal. Chem. 2022, 77, 618–624. [Google Scholar] [CrossRef]
- Scriba, G.K.E. Chiral recognition in separation sciences. Part I: Polysaccharide and cyclodextrin selectors. Trends Anal. Chem. 2019, 120, 115639. [Google Scholar] [CrossRef]
- Fejős, I.; Kalydi, E.; Malanga, M.; Benkovics, G.; Béni, S. Single isomer cyclodextrins as chiral selectors in capillary electrophoresis. J. Chromatogr. A 2020, 1627, 461375. [Google Scholar] [CrossRef] [PubMed]
- Hancu, G.; Papp, L.A.; Tóth, G.; Kelemen, H. The use of dual cyclodextrin chiral selector systems in the enantioseparation of pharmaceuticals by capillary electrophoresis: An overview. Molecules 2021, 26, 2261. [Google Scholar] [CrossRef] [PubMed]
- Peluso, P.; Chankvetadze, B. Native and substituted cyclodextrins as chiral selectors for capillary electrophoresis enantioseparations: Structures, features, application, and molecular modeling. Electrophoresis 2021, 42, 1676–1708. [Google Scholar] [CrossRef] [PubMed]
- Dubský, P.; Dvořák, M.; Ansorge, M. Affinity capillary electrophoresis: The theory of electromigration. Anal. Bioanal. Chem. 2016, 408, 8623–8641. [Google Scholar] [CrossRef]
- Chankvetadze, B.; Blaschke, G. Enantioseparations in capillary electromigration techniques: Recent developments and future trends. J. Chromatogr. A 2001, 906, 309–363. [Google Scholar] [CrossRef]
- Haginaka, J. Enantiomer separation of drugs by capillary electrophoresis using proteins as chiral selectors. J. Chromatogr. A 2000, 874, 235–254. [Google Scholar] [CrossRef]
- Guo, X.; Liu, Q.; Hu, S.; Guo, W.; Yang, Z.; Zhang, Y. Thermodynamic models to elucidate the enantioseparation of drugs with two stereogenic centers by micellar electrokinetic chromatography. J. Chromatogr. A 2017, 1512, 133–142. [Google Scholar] [CrossRef]
- Qi, L.; Qiao, J. Progress of chiral ligand-exchange capillary electrophoresis for enantioseparation. J. Chromatogr. A 2022, 1679, 463381. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, X. Enantioseparation of ofloxacin and its four related substances with ligand exchange-micellar electrokinetic chromatography using copper(II)-L-isoleucine complex as chiral selector. Chirality 2017, 29, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Szabó, Z.I.; Foroughbakhshfasaei, M.; Gál, R.; Horváth, P.; Komjáti, B.; Noszál, B.; Tóth, G. Chiral separation of lenalidomide by liquid chromatography on polysaccharide-type stationary phases and by capillary electrophoresis using cyclodextrin selectors. J. Sep. Sci. 2018, 41, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Fejős, I.; Tóth, G.; Várnai, B.; Szabó, Z.I.; Köteles, I.; Malanga, M.; Béni, S. Enantioseparation of solriamfetol and its major impurity phenylalaninol by capillary electrophoresis using sulfated gamma cyclodextrin. Electrophoresis 2021, 42, 1818–1825. [Google Scholar] [CrossRef]
- Konjaria, M.L.; Scriba, G.K.E. Enantioseparation of analogs of the dipeptide alanyl-phenylalanine by capillary electrophoresis using neutral cyclodextrins as chiral selectors. J. Chromatogr. A 2020, 1623, 461158. [Google Scholar] [CrossRef]
- Kanizsová, L.; Ansorge, M.; Zusková, I.; Dubský, P. Using single-isomer octa(6-O-sulfo)-γ-cyclodextrin for fast capillary zone electrophoretic enantioseparation of pindolol: Determination of complexation constants, software-assisted optimization, and method validation. J. Chromatogr. A 2018, 1568, 214–221. [Google Scholar] [CrossRef]
- Szabó, Z.I.; Ludmerczki, R.; Fiser, B.; Noszál, B.; Tóth, G. Chiral separation of rasagiline using sulfobutylether-β-cyclodextrin: Capillary electrophoresis, NMR and molecular modeling study. Electrophoresis 2019, 40, 1897–1903. [Google Scholar] [CrossRef] [Green Version]
- Szabó, Z.I.; Gál, R.; Szőcs, L.; Ludmerczki, R.; Muntean, D.L.; Noszál, B.; Tóth, G. Validated capillary electrophoretic method for the enantiomeric quality control of R-praziquantel. Electrophoresis 2017, 38, 1886–1894. [Google Scholar] [CrossRef]
- Gogolashvili, A.; Tatunashvili, E.; Chankvetadze, L.; Sohajda, T.; Szeman, J.; Gumustas, M.; Ozkan, S.A.; Salgado, A.; Chankvetadze, B. Separation of terbutaline enantiomers in capillary electrophoresis with cyclodextrin-type chiral selectors and investigation of structure of selector-selectand complexes. J. Chromatogr. A 2018, 1571, 231–239. [Google Scholar] [CrossRef]
- Krait, S.; Salgado, A.; Chankvetadze, B.; Gago, F.; Scriba, G.K.E. Investigation of the complexation between cyclodextrins and medetomidine enantiomers by capillary electrophoresis, NMR spectroscopy and molecular modeling. J. Chromatogr. A 2018, 1567, 198–210. [Google Scholar] [CrossRef]
- Guo, J.; Wang, J.; Lin, H.; Feng, Y.; Shen, H.; Huang, R.; Liu, L.; Zhao, Z. Combination of capillary electrophoresis and molecular modeling to study the enantiomer affinity pattern between β-blockers and anionic cyclodextrin derivatives in a methanolic and water background electrolyte. J. Sep. Sci. 2019, 42, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Michalska, K.; Bocian, W.; Bednarek, E.; Pałys, B.; Cielecka-Piontek, J. Enantioselective recognition of sutezolid by cyclodextrin modified non-aqueous capillary electrophoresis and explanation of complex formation by means of infrared spectroscopy, NMR and molecular modelling. J. Pharm. Biomed. Anal. 2019, 169, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Varga, E.; Benkovics, G.; Darcsi, A.; Várnai, B.; Sohajda, T.; Malanga, M.; Béni, S. Comparative analysis of the full set of methylated β-cyclodextrins as chiral selectors in capillary electrophoresis. Electrophoresis 2019, 40, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Gogolashvili, A.; Lomsadze, K.; Chankvetadze, L.; Takaishvili, N.; Peluso, P.; Dallocchio, R.; Salgado, A.; Chankvetadze, B. Separation of tetrahydrozoline enantiomers in capillary electrophoresis with cyclodextrin-type chiral selectors and investigation of chiral recognition mechanisms. J. Chromatogr. A 2021, 1643, 462084. [Google Scholar] [CrossRef] [PubMed]
- Krait, S.; Salgado, A.; Peluso, P.; Malanga, M.; Sohajda, T.; Benkovics, G.; Naumann, L.; Neusüß, C.; Chankvetadze, B.; Scriba, G.K.E. Complexation of daclatasvir by single isomer methylated β-cyclodextrins studied by capillary electrophoresis, NMR spectroscopy and mass spectrometry. Carbohydr. Polym. 2021, 273, 118486. [Google Scholar] [CrossRef]
- Krait, S.; Salgado, A.; Malanga, M.; Sohajda, T.; Benkovics, G.; Szakály, P.S.; Chankvetadze, B.; Scriba, G.K.E. Structural characterization of methyl-β-cyclodextrins by high-performance liquid chromatography and nuclear magnetic resonance spectroscopy and effect of their isomeric composition on the capillary electrophoresis enantioseparation of daclatasvir. J. Chromatogr. A 2022, 1661, 462675. [Google Scholar] [CrossRef]
- Gogolashvili, A.; Tatunashvili, E.; Chankvetadze, L.; Sohajda, T.; Gumustas, M.; Ozkan, S.A.; Salgado, A.; Chankvetadze, B. Separation of brombuterol enantiomers in capillary electrophoresis with cyclodextrin-type chiral selectors and investigation of structure of selector-selectand complexes using nuclear magnetic resonance spectroscopy. Electrophoresis 2019, 40, 1904–1912. [Google Scholar] [CrossRef]
- Gogolashvili, A.; Chankvetadze, L.; Takaishvili, N.; Salgado, A.; Chankvetadze, B. Separation of terbutaline enantiomers in capillary electrophoresis with neutral cyclodextrin-type chiral selectors and investigation of the structure of selector-selectand complexes using nuclear magnetic resonance spectroscopy. Electrophoresis 2020, 41, 1023–1030. [Google Scholar] [CrossRef]
- Casado, N.; Saz, J.M.; García, M.Á.; Marina, M.L. Modeling-based optimization of the simultaneous enantiomeric separation of multicomponent mixtures of phenoxy acid herbicides using dual cyclodextrin systems by Capillary Electrophoresis. J. Chromatogr. A 2020, 1610, 460552. [Google Scholar] [CrossRef]
- Rizvi, A.S.; Murtaza, G.; Irfan, M.; Xiao, Y.; Qu, F. Determination of kynurenine enantiomers by alpha-cyclodextrin, cationic-βeta-cyclodextrin and their synergy complemented with stacking enrichment in capillary electrophoresis. J. Chromatogr. A 2020, 1622, 461128. [Google Scholar] [CrossRef]
- Chalavi, S.; Fakhari, A.R.; Nojavan, S. Development of a modified partial filling method in capillary electrophoresis using two chiral plugs for the simultaneous enantioseparation of chiral drugs: Comparison with mixed chiral selector capillary electrophoresis. J. Chromatogr. A 2018, 1567, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Fukuyama, T.; Terabe, S. Chiral separation by cyclodextrin-modified micellar electrokinetic chromatography. J. Chromatogr. A 1991, 553, 503–516. [Google Scholar] [CrossRef]
- Yu, R.B.; Quirino, J.P. Bile salts in chiral micellar electrokinetic chromatography: 2000–2020. Molecules 2021, 26, 5531. [Google Scholar] [CrossRef] [PubMed]
- Borst, C.; Holzgrabe, U. Cyclodextrin-mediated enantioseparation in microemulsion electrokinetic chromatography. Methods Mol. Biol. 2013, 970, 363–375. [Google Scholar]
- Greño, M.; Marina, M.L.; Castro-Puyana, M. Enantioseparation by capillary electrophoresis using ionic liquids as chiral selectors. Crit. Rev. Anal. Chem. 2018, 48, 429–446. [Google Scholar] [CrossRef]
- Greño, M.; Marina, M.L.; Castro-Puyana, M. Effect of the combined use of γ-cyclodextrin and a chiral ionic liquid on the enantiomeric separation of homocysteine by capillary electrophoresis. J. Chromatogr. A 2018, 1568, 222–228. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, J.; Xue, S.; Rui, M.; Gao, B.; Li, A.; Bai, J.; Yin, Z.; Anochie, E.M. Enhanced enantioselectivity of native α-cyclodextrins by the synergy of chiral ionic liquids in capillary electrophoresis. J. Sep. Sci. 2018, 41, 4525–4532. [Google Scholar] [CrossRef]
- Zhang, Q. Ionic liquids in capillary electrophoresis for enantioseparation. Trends Anal. Chem. 2018, 100, 145–154. [Google Scholar] [CrossRef]
- Wahl, J.; Holzgrabe, U. Capillary electrophoresis separation of phenethylamine enantiomers using amino acid based ionic liquids. J. Pharm. Biomed. Anal. 2018, 148, 245–250. [Google Scholar] [CrossRef]
- Casado, N.; Salgado, A.; Castro-Puyana, M.; García, M.Á.; Marina, M.L. Enantiomeric separation of ivabradine by cyclodextrin-electrokinetic chromatography. Effect of amino acid chiral ionic liquids. J. Chromatogr. A 2019, 1608, 460407. [Google Scholar] [CrossRef]
- Greño, M.; Salgado, A.; Castro-Puyana, M.; Marina, M.L. Nuclear magnetic resonance to study the interactions acting in the enantiomeric separation of homocysteine by capillary electrophoresis with a dual system of γ-cyclodextrin and the chiral ionic liquid EtCholNTf2. Electrophoresis 2019, 40, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, A.G.; Mavroudi, M.C.; Stavrou, I.J.; Weatherly, C.A.; Kapnissi-Christodoulou, C.P. Synergistic enantioseparation systems with either cyclodextrins or cyclofructans and L-alanine Tert butyl ester lactate. Electrophoresis 2019, 40, 539–546. [Google Scholar] [CrossRef] [PubMed]
- Greño, M.; Castro-Puyana, M.; Marina, M.L. Enantiomeric separation of homocysteine and cysteine by electrokinetic chromatography using mixtures of γ-cyclodextrin and carnitine-based ionic liquids. Microchem. J. 2020, 157, 105070. [Google Scholar] [CrossRef]
- Ren, S.; Xue, S.; Sun, X.; Rui, M.; Wang, L.; Zhang, Q. Investigation of the synergistic effect of chiral ionic liquids as additives in non-aqueous capillary electrophoresis for enantioseparation. J. Chromatogr. A 2020, 1609, 460519. [Google Scholar] [CrossRef]
- Salido-Fortuna, S.; Marina, M.L.; Castro-Puyana, M. Enantiomeric determination of econazole and sulconazole by electrokinetic chromatography using hydroxypropyl-β-cyclodextrin combined with ionic liquids based on L-lysine and L-glutamic acid. J. Chromatogr. A 2020, 1621, 461085. [Google Scholar] [CrossRef] [PubMed]
- Greño, M.; Marina, M.L.; Castro-Puyana, M. Use of single and dual systems of γ-cyclodextrin or γ-cyclodextrin/L-Carnitine derived ionic liquid for the enantiomeric determination of cysteine by electrokinetic chromatography. A comparative study. Microchem. J. 2021, 169, 106596. [Google Scholar] [CrossRef]
- Salido-Fortuna, S.; Fernández-Bachiller, M.I.; Marina, M.L.; Castro-Puyana, M. Synthesis and characterization of carnitine-based ionic liquids and their evaluation as additives in cyclodextrin-electrokinetic chromatography for the chiral separation of thiol amino acids. J. Chromatogr. A 2022, 1670, 462955. [Google Scholar] [CrossRef]
- Li, J.; Yu, T.; Xu, G.; Du, Y.; Liu, Z.; Feng, Z.; Yang, X.; Xi, Y.; Liu, J. Synthesis and application of ionic liquid functionalized β-cyclodextrin, mono-6-deoxy-6-(4-amino-1,2,4-triazolium)-β-cyclodextrin chloride, as chiral selector in capillary electrophoresis. J. Chromatogr. A 2018, 1559, 178–185. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, C.; Chen, J.; Xu, G.; Du, Y.; Ma, X.; Sun, X.; Feng, Z.; Huang, Z. Synthesis and application of tetramethylammonium-carboxymethylated-β-cyclodextrin: A novel ionic liquid in capillary electrophoresis enantioseparation. J. Pharm. Biomed. Anal. 2020, 180, 113030. [Google Scholar] [CrossRef]
- Mu, Y.; Wu, X.; Huang, Y.P.; Liu, Z.S. Investigation of deep eutectic solvents as additives to β-CD for enantiomeric separations of Zopiclone, Salbutamol, and Amlodipine by CE. Electrophoresis 2019, 40, 1992–1995. [Google Scholar] [CrossRef]
- Deng, S.; Pan, J.; Wang, M.; Huang, Y.; Xia, Z. Study on improvement of chiral separation of capillary electrophoresis based on cyclodextrin by deep eutectic solvents. Talanta 2020, 220, 121419. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ren, S.; Teng, C.; Liu, H.; Zhang, Q. The role of deep eutectic solvents in chiral capillary electrokinetic chromatography: A comparative study based on α-cyclodextrin chiral selector. J. Mol. Liq. 2022, 359, 119281. [Google Scholar] [CrossRef]
- Wren, S.A.C. Theory of chiral separation in capillary electrophoresis. J. Chromatogr. 1993, 636, 57–62. [Google Scholar] [CrossRef]
- Pálmarsdóttir, S.; Edholm, L.-E. Capillary electrophoresis for separation of drug enantiomers using cyclodextrins as chiral selectors: Influence of experimental parameters on separation. J. Chromatogr. A 1994, 666, 337–350. [Google Scholar] [CrossRef]
- Williams, B.A.; Vigh, G. Dry look at the CHARM (charged resolving agent migration) model of enantiomer separations by capillary electrophoresis. J. Chromatogr. A 1997, 777, 295–309. [Google Scholar] [CrossRef]
- Dejaegher, B.; Mangelings, D.; Heyden, Y.V. Experimental design methodologies in the optimization of chiral CE or CEC separations: An overview. Methods Mol. Biol. 2013, 970, 409–427. [Google Scholar]
- Wahl, O.; Holzgrabe, U. Evaluation of enantiomeric purity of magnesium-L-aspartate dihydrate. J. Pharm. Biomed. Anal. 2015, 102, 100–109. [Google Scholar] [CrossRef]
- Kazsoki, A.; Fejős, I.; Sohajda, T.; Zhou, W.; Hu, W.; Szente, L.; Béni, S. Development and validation of a cyclodextrin-modified capillary electrophoresis method for the enantiomeric separation of vildagliptin enantiomers. Electrophoresis 2016, 37, 1318–1325. [Google Scholar] [CrossRef]
- Flor, S.; Huala Juan, M.; Tripodi, V.; Lucangioli, S. Development of an enantioselective capillary electrophoretic method for the simultaneous determination of montelukast enantiomeric and diastereoisomeric forms and its main degradation product. Electrophoresis 2016, 37, 2420–2428. [Google Scholar] [CrossRef]
- Meng, R.; Kang, J. Determination of the stereoisomeric impurities of sitafloxacin by capillary electrophoresis with dual chiral additives. J. Chromatogr. A 2017, 1506, 120–127. [Google Scholar] [CrossRef]
- Papp, L.A.; Hancu, G.; Gyéresi, Á.; Kelemen, H.; Szabó, Z.-I.; Noszál, B.; Dubský, P.; Tóth, G. Chiral separation of lansoprazole and rabeprazole by capillary electrophoresis using dual cyclodextrin systems. Electrophoresis 2019, 40, 2799–2805. [Google Scholar] [CrossRef] [PubMed]
- Budău, M.; Hancu, G.; Muntean, D.L.; Papp, L.A.; Cârje, A.G.; Garaj, V. Enantioseparation of citalopram enantiomers by capillary electrophoresis: Method development through experimental design and computational modelling. Chirality 2020, 32, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Mai, X.L.; Pham, T.V.; Le, T.A.; Nguyen, B.T.; Nguyen, N.V.T.; Kang, J.S.; Mar, W.; Kim, K.H. A capillary electrophoresis method for the determination of the linagliptin enantiomeric impurity. J. Sep. Sci. 2020, 43, 4480–4487. [Google Scholar] [CrossRef] [PubMed]
- Orlandini, S.; Pasquini, B.; Del Bubba, M.; Pinzauti, S.; Furlanetto, S. Quality by design in the chiral separation strategy for the determination of enantiomeric impurities: Development of a capillary electrophoresis method based on dual cyclodextrin systems for the analysis of levosulpiride. J. Chromatogr. A 2015, 1380, 177–185. [Google Scholar] [CrossRef]
- Krait, S.; Douša, M.; Scriba, G.K.E. Quality by Design-guided development of a capillary electrophoresis method for the chiral purity determination of ambrisentan. Chromatographia 2016, 79, 1343–1350. [Google Scholar] [CrossRef]
- Orlandini, S.; Pasquini, B.; Caprini, C.; Del Bubba, M.; Douša, M.; Pinzauti, S.; Furlanetto, S. Enantioseparation and impurity determination of ambrisentan using cyclodextrin-modified micellar electrokinetic chromatography: Visualizing the design space within quality by design framework. J. Chromatogr. A 2016, 1467, 363–371. [Google Scholar] [CrossRef]
- Niedermeier, S.; Scriba, G. A Quality by Design-based approach to a capillary electrokinetic assay for the determination of dextromepromazine and levopromazine sulfoxide as impurities of levopromazine. J. Pharm. Biomed. Anal. 2017, 146, 402–409. [Google Scholar] [CrossRef]
- Krait, S.; Heuermann, M.; Scriba, G.K.E. Development of a capillary electrophoresis method for the determination of the chiral purity of dextromethorphan by a dual selector system using quality by design methodology. J. Sep. Sci. 2018, 41, 1405–1413. [Google Scholar] [CrossRef]
- Harnisch, H.; Chien, Y.; Scriba, G.K.E. Capillary electrophoresis method for the chiral purity determination of pregabalin derivatized with dansyl chloride. Chromatographia 2018, 81, 719–725. [Google Scholar] [CrossRef]
- Krait, S.; Scriba, G.K.E. Quality by design-assisted development of a capillary electrophoresis method for the chiral purity determination of dexmedetomidine. Electrophoresis 2018, 39, 2575–2580. [Google Scholar] [CrossRef]
- Pasquini, B.; Orlandini, S.; Villar-Navarro, M.; Caprini, C.; Del Bubba, M.; Douša, M.; Giuffrida, A.; Gotti, R.; Furlanetto, S. Chiral capillary zone electrophoresis in enantioseparation and analysis of cinacalcet impurities: Use of Quality by Design principles in method development. J. Chromatogr. A 2018, 1568, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Harnisch, H.; Scriba, G.K.E. Capillary electrophoresis method for the determination of (R)-dapoxetine, (3S)-3-(dimethylamino)-3-phenyl-1-propanol, (S)-3-amino-3-phenyl-1-propanol and 1-naphthol as impurities of dapoxetine hydrochloride. J. Pharm. Biomed. Anal. 2019, 162, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Niedermeier, S.; Scriba, G.K.E. Quality by Design-based development of a chiral capillary electrophoresis method for the determination of dextrodropropizine and 1-phenylpiperazine as impurities of levodropropizine. Chromatographia 2020, 83, 123–129. [Google Scholar] [CrossRef]
- Niedermeier, S.; Scriba, G.K.E. Chiral separation of four phenothiazines by nonaqueous capillary electrophoresis and quality by design-based method development for quantification of dextromepromazine as chiral impurity of levomepromazine. J. Chromatogr. A 2020, 1624, 461232. [Google Scholar] [CrossRef]
- Krait, S.; Schneidmadel, F.R.; Scriba, G.K.E. Quality by design-assisted development of a capillary electrophoresis method for the enantiomeric purity determination of tenofovir. Electrophoresis 2022, 43, 964–969. [Google Scholar] [CrossRef]
- ICH Harmonised Tripartite Guideline. Impurities in New Drug Substances Q3A(R2); International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use: Geneva, Switzerland, 2006. [Google Scholar]
- Orlandini, S.; Gotti, R.; Furlanetto, S. Multivariate optimization of capillary electrophoresis methods: A critical review. J. Pharm. Biomed. Anal. 2014, 87, 290–307. [Google Scholar] [CrossRef]
- Montgomery, D.C. Design and Analysis of Experiments, 4th ed.; John Wiley & Sons: New York, NY, USA, 1997. [Google Scholar]
- Perovani, I.S.; Serpellone, C.O.; de Oliveira, A.R.M. An appraisal of experimental designs: Application to enantioselective capillary electromigration techniques. Electrophoresis 2021, 42, 1726–1743. [Google Scholar] [CrossRef]
- Herrador, M.A.; Asuero, A.G.; Gonzalez, A.G. Estimation of the uncertainty of indirect measurements from the propagation of distributions by using the Monte-Carlo method: An overview. Chemom. Intell. Lab. Syst. 2005, 79, 115–122. [Google Scholar] [CrossRef]
- Peraman, R.; Bhadraya, K.; Padmanabha Reddy, Y. Analytical quality by design: A tool for regulatory flexibility and robust analytics. Int. J. Anal. Chem. 2015, 2015, 868727. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CE Technique | Pharmaceuticals | Matrix | DoE Methodology Factors/Responses | Optimized Analytical Conditions | Reference |
|---|---|---|---|---|---|
| CZE-LIF | Magnesium-l-aspartate (chiral impurity: d-aspartic acid) | bulk substance, tablets | Screening FFD BGE concentration, BGE pH, HP-β-CD concentration/chiral resolution D-aspartic acid impurity determination at 0.06% | 50 mM phosphate BGE, pH 7.0, 18 mM HP-β-CD, 18 mM (v/v) DMSO, 20 kV, 25 °C | [122] |
| CZE-UV | Vildagliptin (chiral impurity: R-vildagliptin) | bulk substance, tablets | OD BGE concentration, BGE pH, SBE-α-CD concentration, temperature, voltage, injection parameters/chiral resolution Robustness testing: PBD R-vildagliptin impurity determination at 0.43% level | 75 mM Tris-acetate BGE, pH 4.75, 20 mM SBE-α-CD, 25 kV, 15 °C, 200 nm | [123] |
| MEKC-UV | Montelukast (chiral impurities: R,R-trans-montelukast, R,S-cis-montelukast, S,R-cis-montelukast) | bulk substance, chewable tablets, oral granules | OFAT BGE concentration, TM-γ-CD concentration, SBE-β-CD concentration, temperature Screening FFD BGE pH, voltage/chiral resolution Robustness testing: PBD R,R-trans-montelukast impurity determination at 0.02% level | 20 mM borate BGE, 10 mM SDS, pH 9.0, 10 mM TM-γ-CD, 10 mM SBE-β-CD, 18 kV, 15 °C, 254 nm | [124] |
| CZE-UV | Sitafloxacin (chiral impurities: R,R,S-sitafloxacin, S,S,R-sitafloxacin, R,S,R-sitafloxacin) | bulk substance | Screening FrFD BGE concentration, BGE pH, γ-CD concentration, Cu2+ concentration, d-phenylalanine concentration, temperature, voltage Optimization FCCD BGE pH, γ-CD concentration, Cu2+ concentration, d-phenylalanine concentration/chiral resolution, migration time Robustness testing: PBD R,S,R-sitafloxacin enantiomeric impurity determination at 0.1% level | 15 mM phosphate BGE, pH 4.5, 15 mM d-phenylalanine, 20 mM CuSO4, 20 mM γ-CD, 15 kV, 25 °C, 297 nm | [125] |
| CZE-UV | Lenalidomide (chiral impurity: R-lenalidomide) | bulk substance | Optimization FCCD BGE concentration, temperature, voltage/chiral resolution R-lenalidomide impurity determination at 0.1% level | 30 mM phosphate BGE, pH 6.5, 30 mM SBE-β-CD, 12 kV, 10 °C, 210 nm | [78] |
| CZE-UV | R-lansoprazole (chiral impurity S-lansoprazole), R-rabeprazole (chiral impurity S-rabeprazole) | bulk substance, capsules | Screening FrFD BGE concentration, BGE pH, SBE-β-CD concentration, γ-CD concentration, temperature, voltage Optimization CCD SBE-β-CD concentration, voltage, temperature/chiral resolution, migration time Robustness testing: PBD R-lansoprazole and S-rabeprazole enantiomeric impurity determination at 0.15% level | Lansoprazole: 25 mM phosphate BGE, pH 7.0, 10 mM SBE-β-CD, 20 mM γ-CD, 20 kV, 17 °C, 210 nm Rabeprazole: 25 mM phosphate BGE, pH 7.0, 15 mM SBE-β-CD, 30 mM γ-CD, 20 kV, 18 °C, 210 nm | [126] |
| CZE-UV | S-citalopram (chiral impurity R-citalopram) | bulk substance, tablets | Screening FrFD BGE concentration, BGE pH, CM-β-CD concentration, temperature, voltage, injection pressure Optimization FCCD CM-β-CD concentration, temperature, voltage/chiral resolution, migration time Robustness testing: PBD R-citalopram enantiomeric impurity determination at 0.05% level | 25 mM phosphate BGE, pH 7.0, 3 mM CM-β-CD, 15 kV, 17.5 °C, 230 nm | [127] |
| CZE-UV | Linagliptin (chiral impurity S-linagliptin) | bulk substance | Screening FrFD BGE concentration, BGE pH, CM-β-CD concentration, temperature, voltage, injection time/chiral resolution, resolution between the internal standard and chiral impurity, migration time, peak height, distance between linagliptin peak and EOF Optimization I-Optimal design BGE concentration, BGE pH, CM-β-CD concentration, temperature, voltage/chiral resolution, migration time, peak height, distance between linagliptin peak and EOF Robustness testing: PBD S-linagliptin enantiomeric impurity determination at 0.05% level | 70 mM sodium acetate BGE, pH 6.10, 4.7 mM CM-β-CD, 28 kV, 25 °C, 200 nm | [128] |
| CZE-UV | R-solriamfetol (chiral impurities: S-solriamfetol, R-phenylalaninol, S-phenylalaninol) | bulk substance, tablets | Screening FrFD BGE concentration, BGE pH, S-γ-CD concentration, temperature, voltage/chiral resolution, migration time, resolution between phenylalaninol enantiomers, resolution between S-solriamfetol enantiomeric impurity and R-phenylalaninol S-solriamfetol enantiomeric impurity determination at 0.1% level | 45 mM Tris-acetate BGE, pH 4.5, 4 mM S-γ-CD, 19.5 kV, 21 °C, 200 nm | [79] |
| CE Technique | Pharmaceuticals | Matrix | DoE Methodology Factors/Responses | Design Space Determination/Validation/Optimized Analytical Conditions | References |
|---|---|---|---|---|---|
| CZE-UV | Levosulpiride (chiral impurity dextrosulpiride) | bulk substance, injection solutions | Asymmetric screening matrix BGE concentration, BGE pH, neutral CD concentration, type of neutral CD, S-β-CD concentration, voltage Optimization DD BGE pH, M-β-CD concentration, S-β-CD concentration, voltage/chiral resolution, migration time | Monte Carlo simulation Robustness testing: PBD R-sulpiride enantiomeric impurity determination at 0.1% level 5 mM Britton-Robinson BGE, pH 3.45, 10 mM S-β-CD, 34 mM M-β-CD, −14 kV, 16 °C, 214 nm | [129] |
| CZE-UV | S-Ambrisentan (chiral impurity R-ambrisentan) | bulk substance, real sample | Screening FrFD BGE concentration, BGE pH, γ-CD concentration, temperature, voltage Optimization FCCD BGE concentration, temperature, voltage/chiral resolution, migration time | Monte Carlo simulation Robustness testing: PBD R-ambrisentan enantiomeric impurity determination at 0.1% level 50 mM sodium acetate BGE, pH 4.0, 30 mM γ-CD, 25 kV, 25 °C, 200 nm | [130] |
| MEKC-UV | S-Ambrisentan (chiral impurity R-ambrisentan and three achiral impurities) | bulk substance, coated tablets | Asymmetric screening matrix BGE concentration, BGE pH, γ-CD concentration, SDS concentration, temperature, voltage, capillary length Optimization FCCD BGE pH, γ-CD concentration, voltage/chiral resolution, migration time | Monte Carlo simulation Robustness testing: PBD R-ambrisentan enantiomeric impurity determination at 0.1% level 100 mM borate BGE, pH 9.20, 100 mM SDS, 50 mM γ-CD, 30 kV, 22 °C, 200 nm | [131] |
| CZE-UV | Levomepromazine (chiral impurity dextromepromazine, levomepromazine sulphoxide diastereomers) | bulk substance, injection solution | Screening FrFD BGE concentration, BGE pH, HP-γ-CD concentration, temperature, voltage Optimization FCCD BGE concentration, BGE pH, HP-γ-CD concentration/chiral resolution expressed as selectivity, resolution, migration time | Monte Carlo simulation Robustness testing: PBD Dextromepromazine enantiomeric impurity determination in the 0.1–1.0% range (0.25 mg/mL levomepromazine) 100 mM citric acid BGE, pH 2.85, 3.6 mg/mL HP-γ-CD, 25 kV, 15 °C, 253 nm | [132] |
| CZE-UV | Dextromethorphan (chiral impurity levomethorphan) | bulk substance, capsules | Screening FrFD BGE concentration, BGE pH, M-α-CD concentration, S-β-CD concentration, temperature, voltage Optimization FCCDM-α-CD concentration, S-β-CD concentration, voltage/migration time, number of theoretical plates, height of levomethorphan, tailing factor of dextromethorphan | Monte Carlo simulation Robustness testing: PBD Levomethorphan enantiomeric impurity determination at 0.1% level 30 mM phosphate BGE, pH 6.5, 16 mg/mL S-β-CD, 14 mg/mL M-α-CD, 20 kV, 20 °C, 200 nm | [133] |
| CZE-UV (derivatization with dansyl chloride) | Pregabalin (chiral impurity R-pregabalin) | bulk substance, capsules | Screening D-optimal design BGE concentration, BGE pH, TM-β-CD concentration, temperature, voltage Optimization FCCD TM-β-CD concentration, temperature, voltage/chiral resolution, migration time | Monte Carlo simulation Robustness testing: PBD R-pregabalin enantiomeric impurity determination at 0.05% level 100 mM phosphate BGE, pH 2.5, 40 mg/mL TM-β-CD, 15 kV, 25 °C, 220 nm | [134] |
| CZE-UV | Dexmedetomidine (chiral impurity levomedetomidine) | bulk substance, tablets | Screening FrFD BGE concentration, BGE pH, S-β-CD concentration, temperature, voltage Optimization FCCD S-β-CD concentration, temperature, voltage/chiral resolution expressed as selectivity, migration time, current | Monte Carlo simulation Robustness testing: PBD Levomedetomidine enantiomeric impurity determination at 0.1% level 50 mM phosphate BGE, pH 6.5, 40 mg/mL S-β-CD, 10 kV, 17 °C, 200 nm | [135] |
| CZE-UV | Cinacalcet (chiral impurity S-cinacalcet and two chiral impurities) | bulk substance, tablets | Optimization BBD BGE pH, HP-γ-CD concentration, methanol concentration, voltage/chiral resolution, migration time | Monte Carlo simulation Robustness testing: PBD S-cinacalcet enantiomeric impurity determination at 0.1% level 150 mM phosphate BGE, pH 2.7, 3.1 mM HP-γ-CD, 2% (v/v) methanol, 26 kV, 18 °C, 220 nm | [136] |
| CZE-UV | S-dapoxetine (chiral impurities: R-dapoxetine, (S)-3-amino-3-phenylpropan-1-ol, (3S)-3-(dimethylamino)-3-phenylpropan-1-ol) | bulk substance, tablets | Screening FrFD BGE concentration, BGE pH, CD concentration (DM-β-CD: S-γ-CD 1:1), temperature, voltage Optimization FCCD DM-β-CD concentration, S-γ-CD concentration, voltage/chiral resolution, migration time, peak symmetry, current | Monte Carlo simulation Robustness testing: PBD R-dapoxetine enantiomeric impurity determination in the 0.05–1.0% range 50 mM phosphate BGE, pH 6.3, 40.2 mg/mL DM-β-CD, 45 mg/mL S-γ-CD, 9 kV, 15 °C, 215 nm | [137] |
| CZE-UV | Levodropropizine (chiral impurity dextrodropropizine and achiral precursor impurity) | bulk substance, pharmaceutical drops | Screening FFD S-β-CD concentration, 2-propanol concentration, temperature, voltage Optimization FCCD S-β-CD concentration, temperature/separation factors, migration time, current | Monte Carlo simulation Robustness testing: PBD dextrodropropizine enantiomeric impurity determination at 0.5% level 25 mM phosphate BGE, pH 7.0, 23.5 mg/mL S-β-CD, 10% (v/v) 2-propanol, 16.5 kV, 16.3 °C, 200 nm | [138] |
| NACE-UV | Levomepromazine (chiral impurity dextromepromazine) | bulk substance, tablets | Screening FrFD ammonium acetate concentration, acetic acid concentration, HDMS-β-CD concentration, temperature, voltage/separation factor, migration time, current Optimization FCCD ammonium acetate concentration, HDMS-β-CD concentration, voltage/chiral resolution, migration time, separation factors | Monte Carlo simulation Robustness testing: PBD dextromepromazine enantiomeric impurity determination in the 0.01–3.0% range (0.74 mg/mL levomepromazine) 75 mM acetic acid, 55 mM ammonium acetate in methanol BGE, 27.5 mg/mL HDMS-β-CD, 22 kV, 15 °C, 250 nm | [139] |
| CZE-UV | Tenofovir (chiral impurity S-tenofovir) | bulk substance | Screening FrFD BGE concentration, BGE pH, QA-β-CD concentration, temperature, voltage Optimization FCCD BGE pH, temperature, voltage/migration time, separation factor, current | Monte Carlo simulation Robustness testing: PBD S-tenofovir enantiomeric impurity determination at 0.1% level 100 mM phosphate BGE, pH 6.4, 45 mg/mL QA-β-CD, 18 kV, 22 °C, 257 nm | [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orlandini, S.; Hancu, G.; Szabó, Z.-I.; Modroiu, A.; Papp, L.-A.; Gotti, R.; Furlanetto, S. New Trends in the Quality Control of Enantiomeric Drugs: Quality by Design-Compliant Development of Chiral Capillary Electrophoresis Methods. Molecules 2022, 27, 7058. https://doi.org/10.3390/molecules27207058
Orlandini S, Hancu G, Szabó Z-I, Modroiu A, Papp L-A, Gotti R, Furlanetto S. New Trends in the Quality Control of Enantiomeric Drugs: Quality by Design-Compliant Development of Chiral Capillary Electrophoresis Methods. Molecules. 2022; 27(20):7058. https://doi.org/10.3390/molecules27207058
Chicago/Turabian StyleOrlandini, Serena, Gabriel Hancu, Zoltán-István Szabó, Adriana Modroiu, Lajos-Attila Papp, Roberto Gotti, and Sandra Furlanetto. 2022. "New Trends in the Quality Control of Enantiomeric Drugs: Quality by Design-Compliant Development of Chiral Capillary Electrophoresis Methods" Molecules 27, no. 20: 7058. https://doi.org/10.3390/molecules27207058