
UHPLC Enantiomer Resolution for the ɑ/β-Adrenoceptor Antagonist R/S-Carvedilol and Its Major Active Metabolites on Chiralpak IB N-5



Abstract
:1. Introduction
2. Results
2.1. Design of Experiment Approach for Enantiomer Resolution of CAR and Its Metabolites
2.1.1. Screening Including Enantiomer Separation of the Most Critical Analyte (4′-OHC) and Total Run-Time (All Analytes) as the Two Optimizable Measures
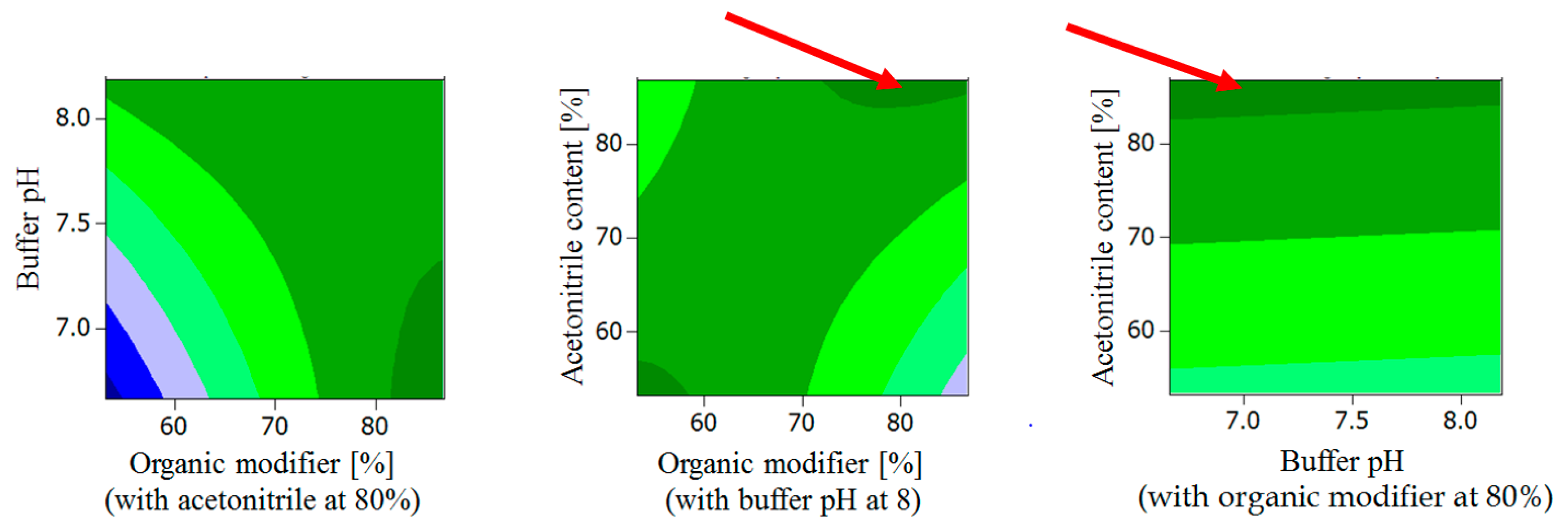
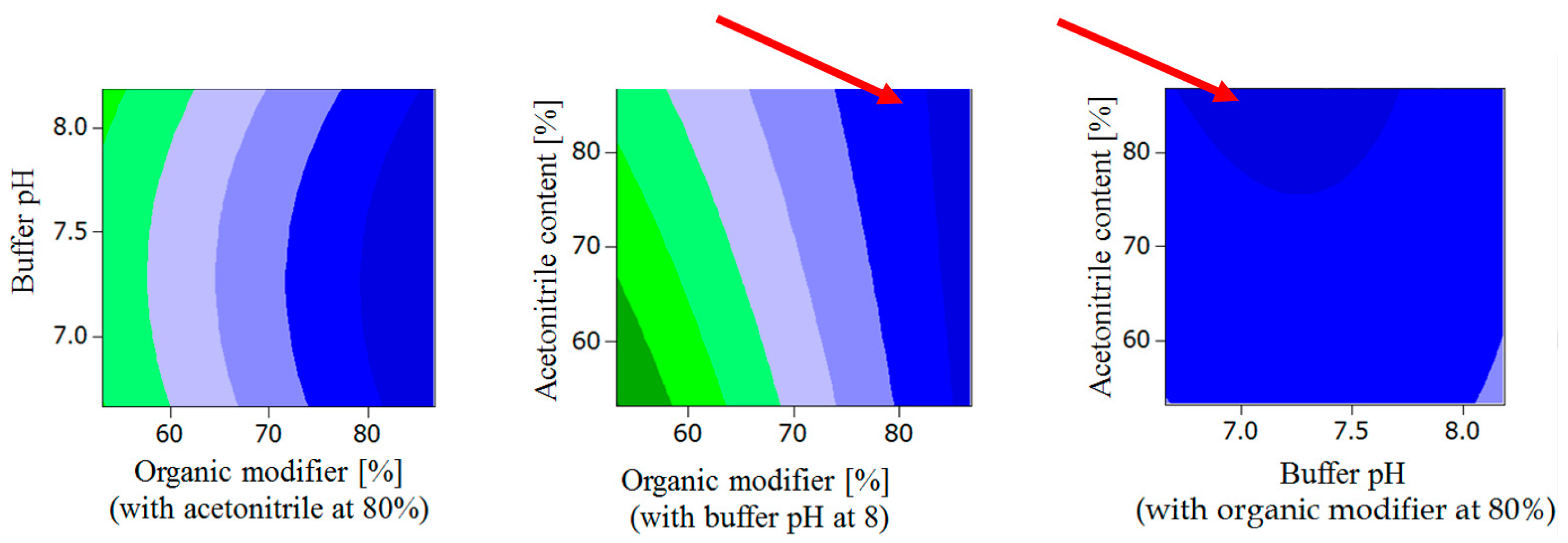
2.1.2. General Design for the Optimization of Conditions and Respective Outcomes
- Buffer pH < 7.3, organic modifier > 80% with acetonitrile content in organic modifier at 80 %.
- Organic modifier < 55% with acetonitrile content in organic modifier < 55%, Buffer pH at 8.
- Organic modifier > 85% with acetonitrile content in organic modifier > 70%, Buffer pH at 8.
- Organic modifier > 85% with acetonitrile content in organic modifier at 80%, Buffer pH 6.5–8.5.
- Acetonitrile content in organic modifier range 60–80%, organic modifier > 80%, Buffer pH at 8.
- Buffer pH range 6.5 to 8.2, organic modifier > 80% with acetonitrile in organic modifier at 80%.
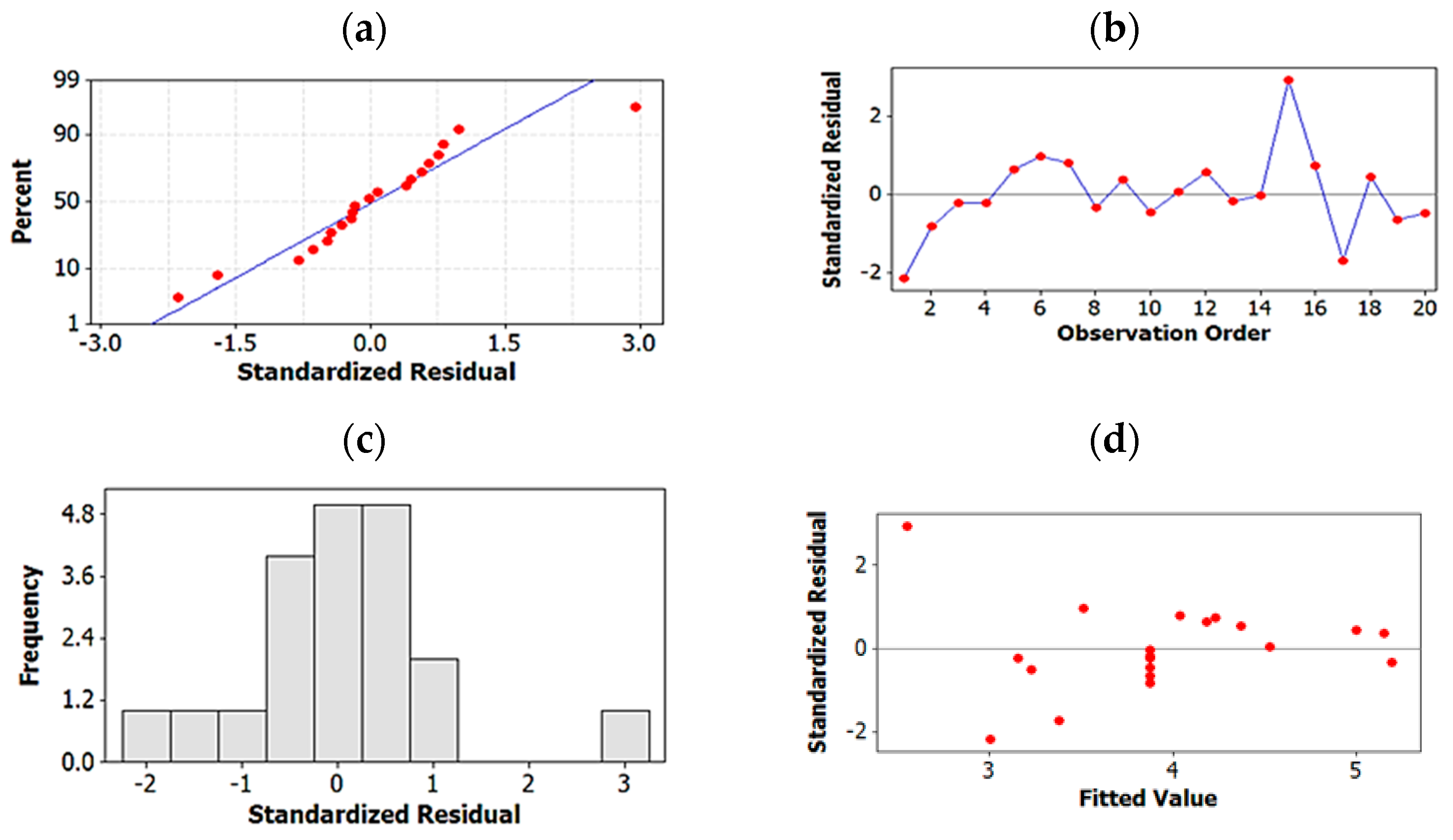
2.1.3. Regression Analysis for Further Optimization and for Elimination of Insignificant Terms
- Rs = Resolution
- X1 = Percent of organic modifier
- X2 = Buffer pH
- X3 = Acetonitrile percentage of organic phase
- t’R = Adjusted retention time
- X1 = Percent of organic modifier
- X2 = Buffer pH
- X3 = Acetonitrile percentage in organic modifier
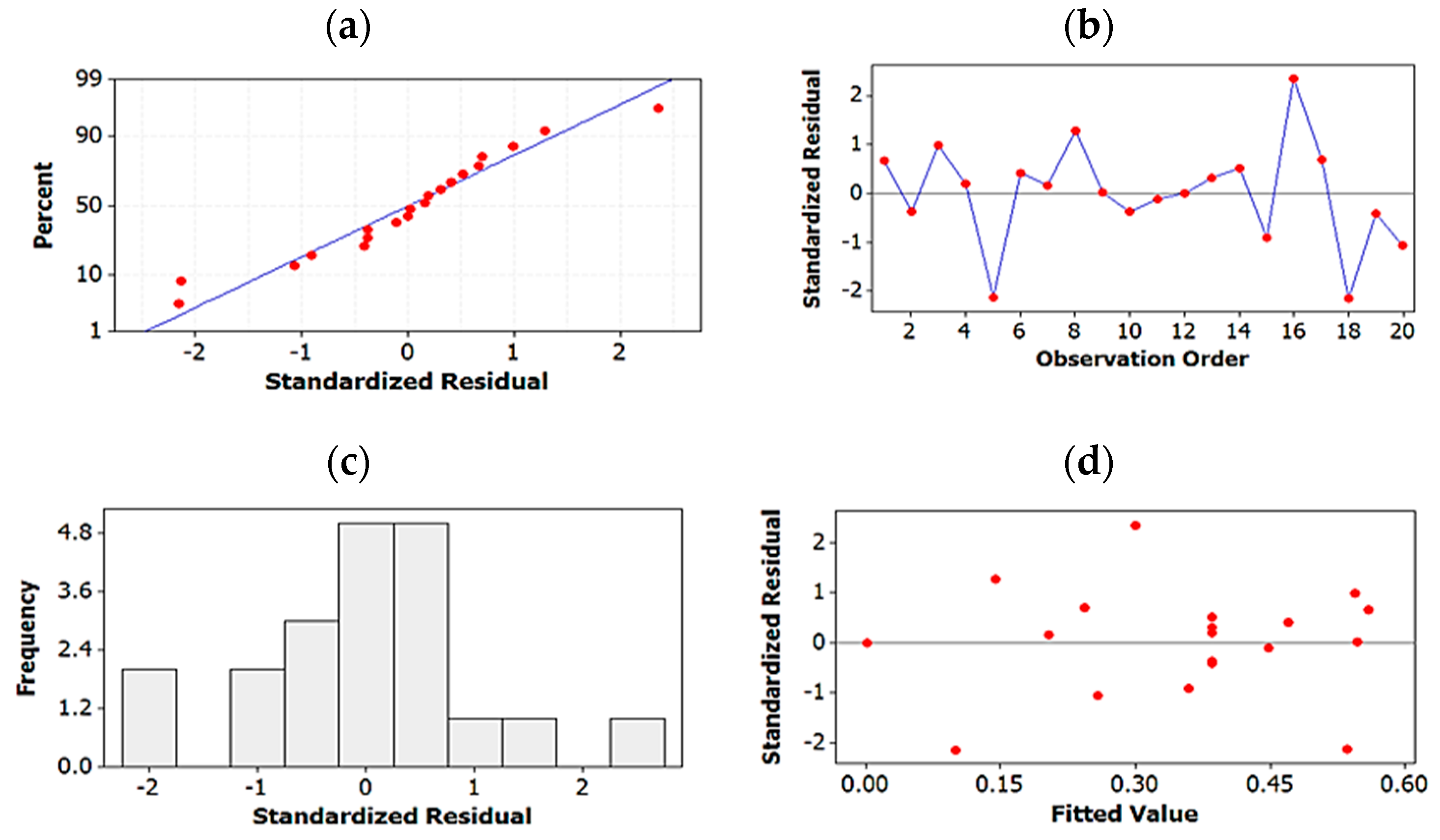
2.1.4. Testing Model Predictability
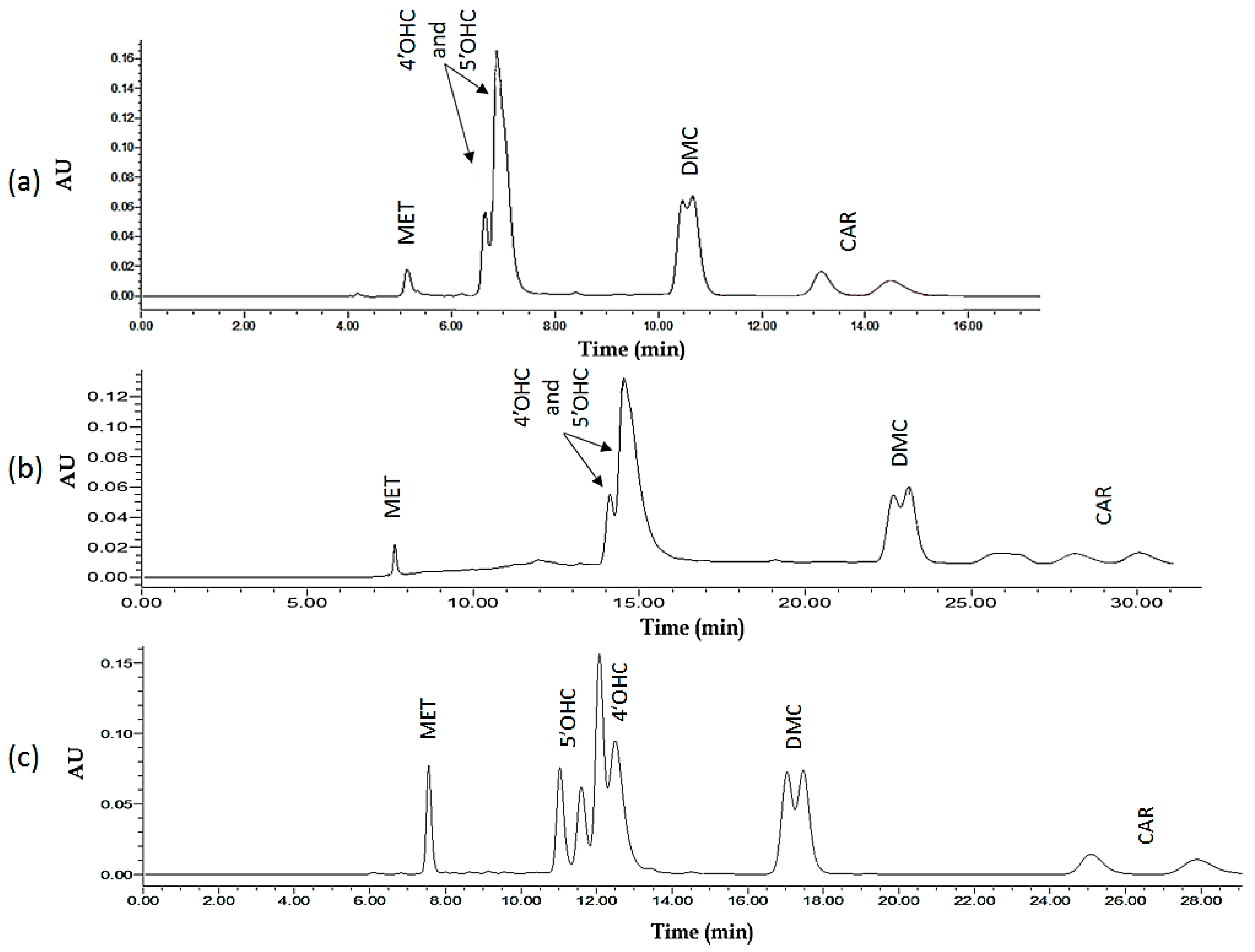
2.1.5. Global Solution: Assay Method Optimized on the Basis of Resolution and Total Run-Time
2.2. CSP Regeneration and Validation of a Preliminary Plasma Assay
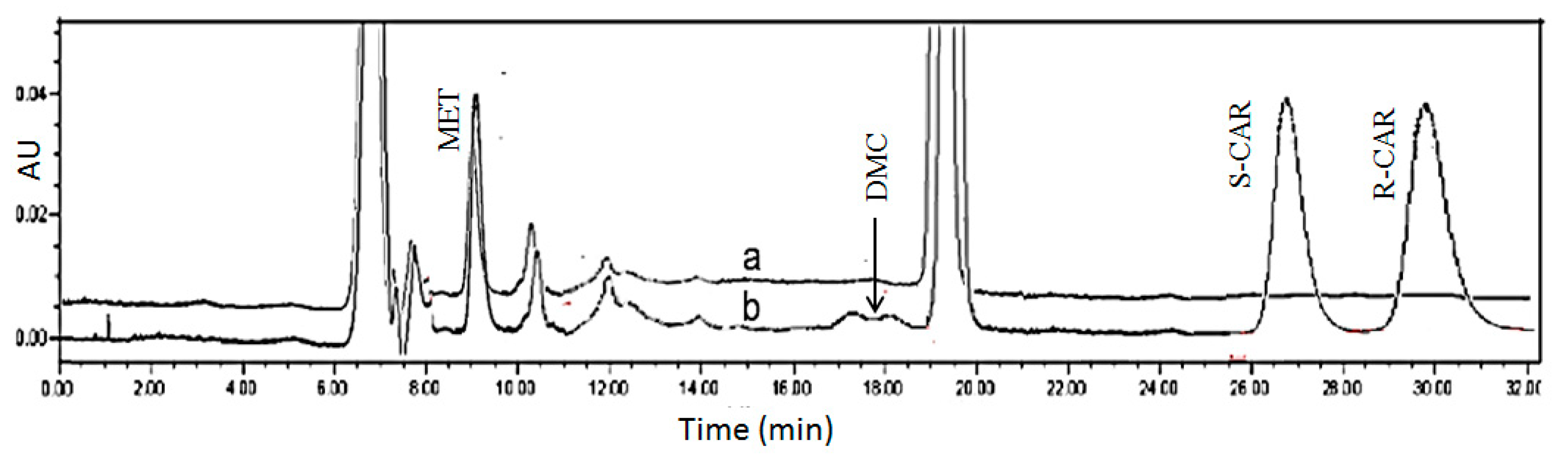
2.2.1. CSP Regeneration
2.2.2. Validation of Plasma Assay
2.3. Assay Application to Patient Plasma Samples
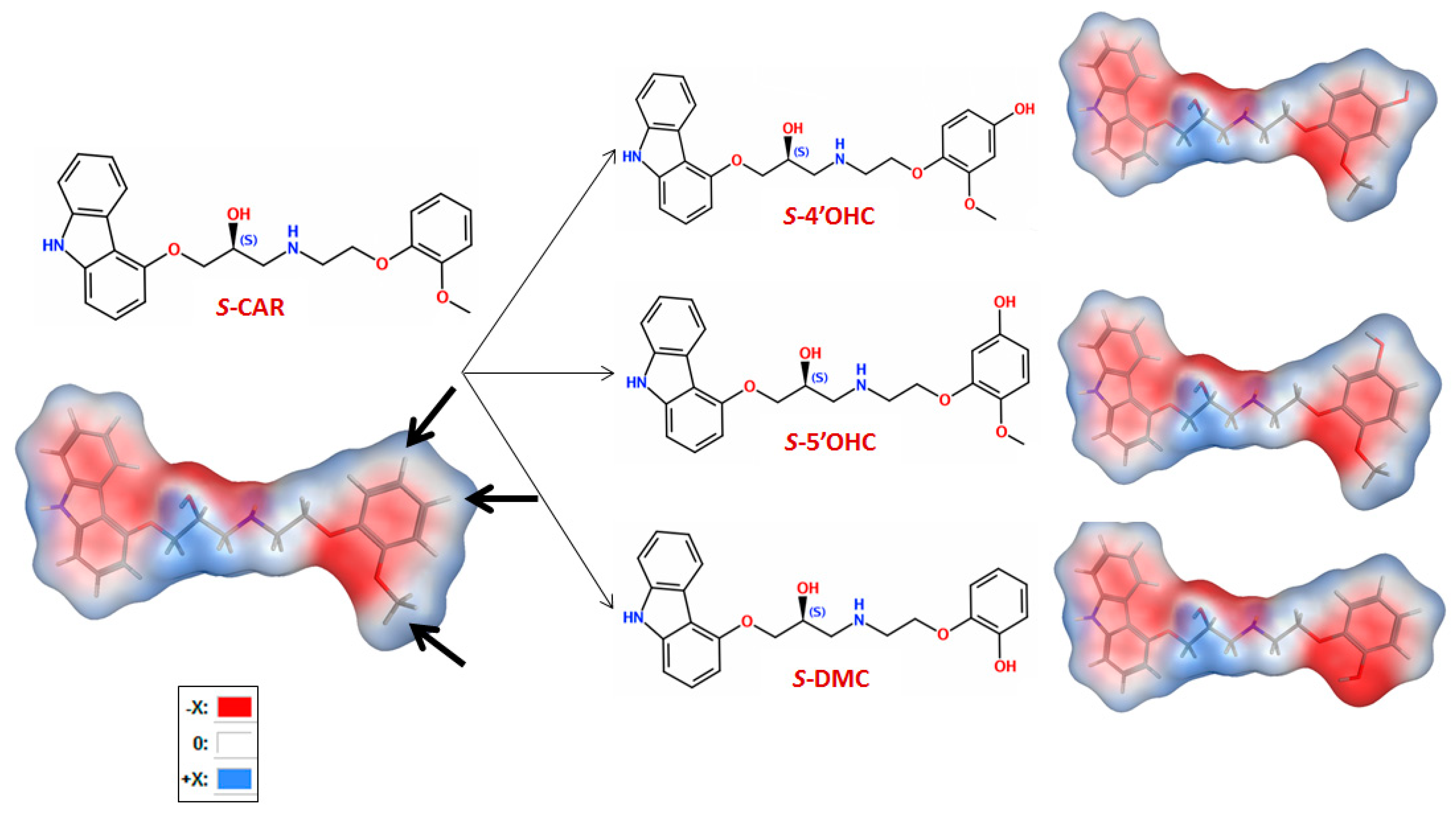
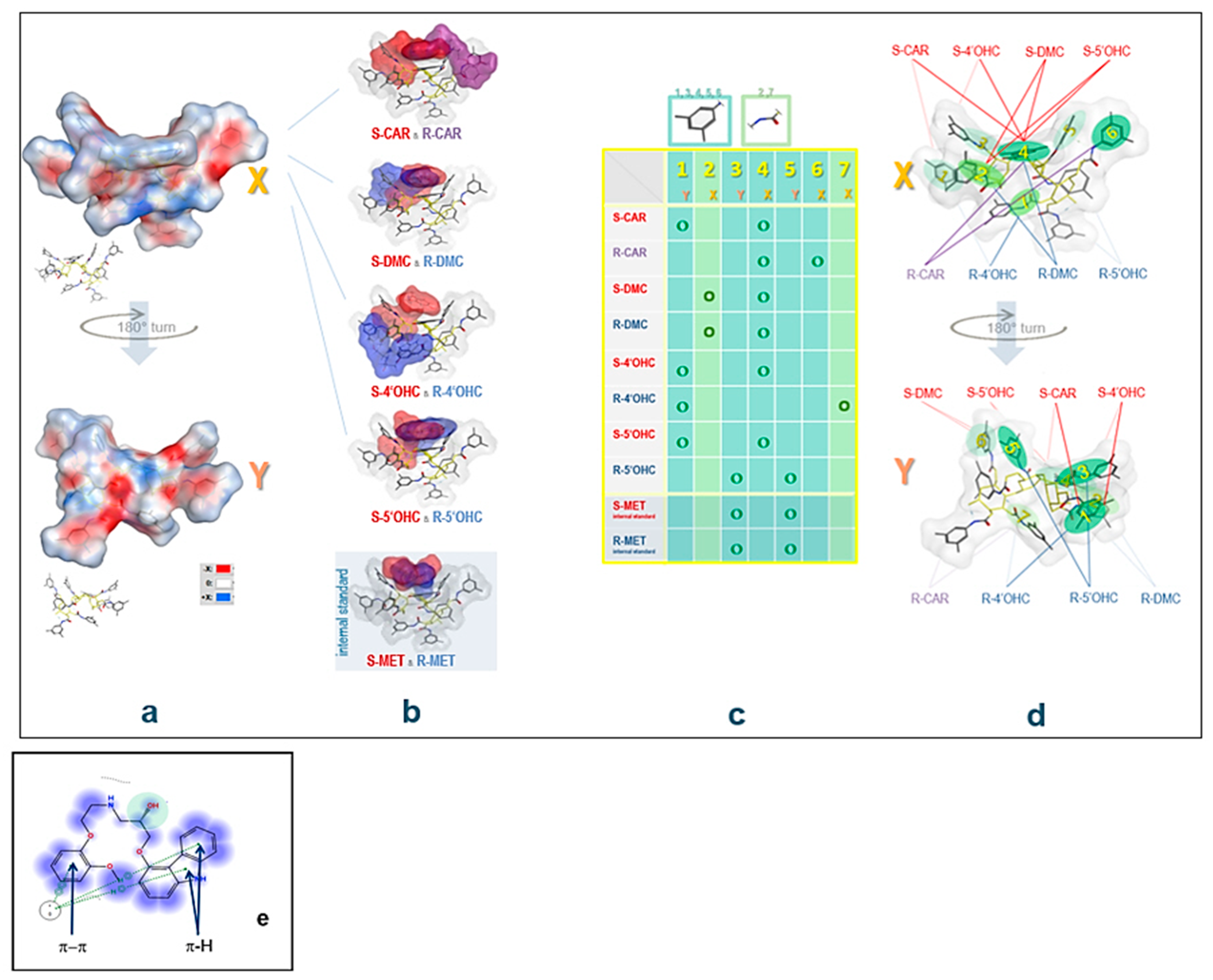
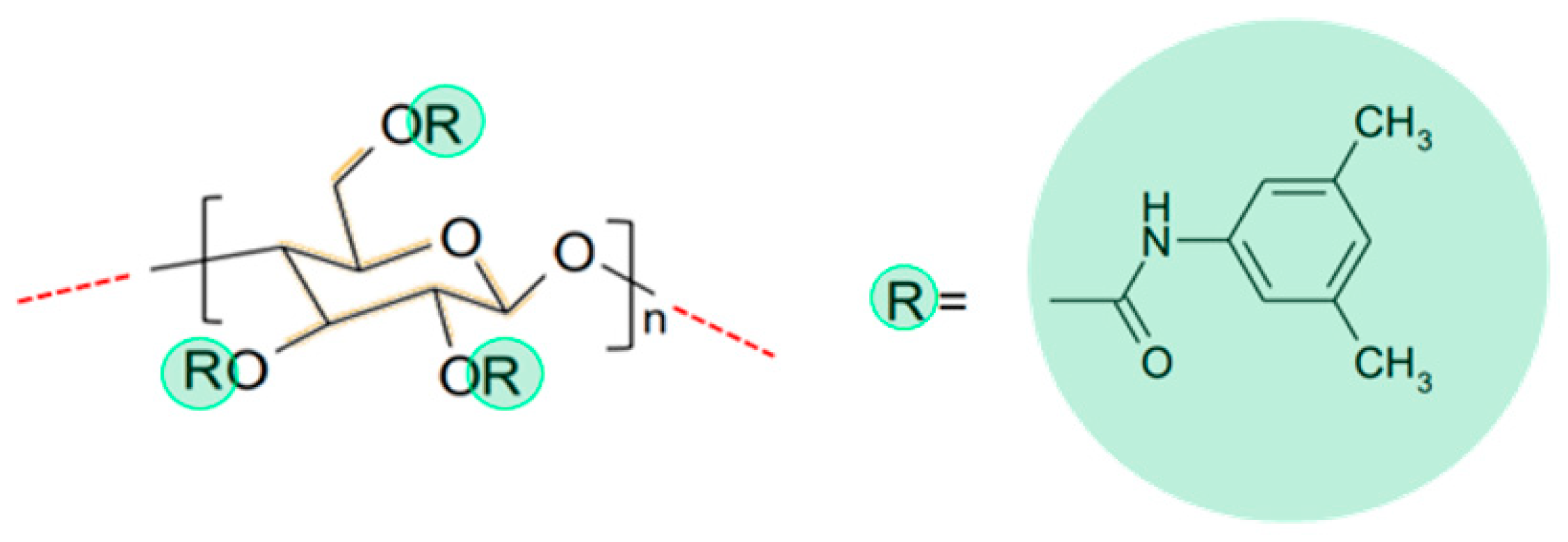
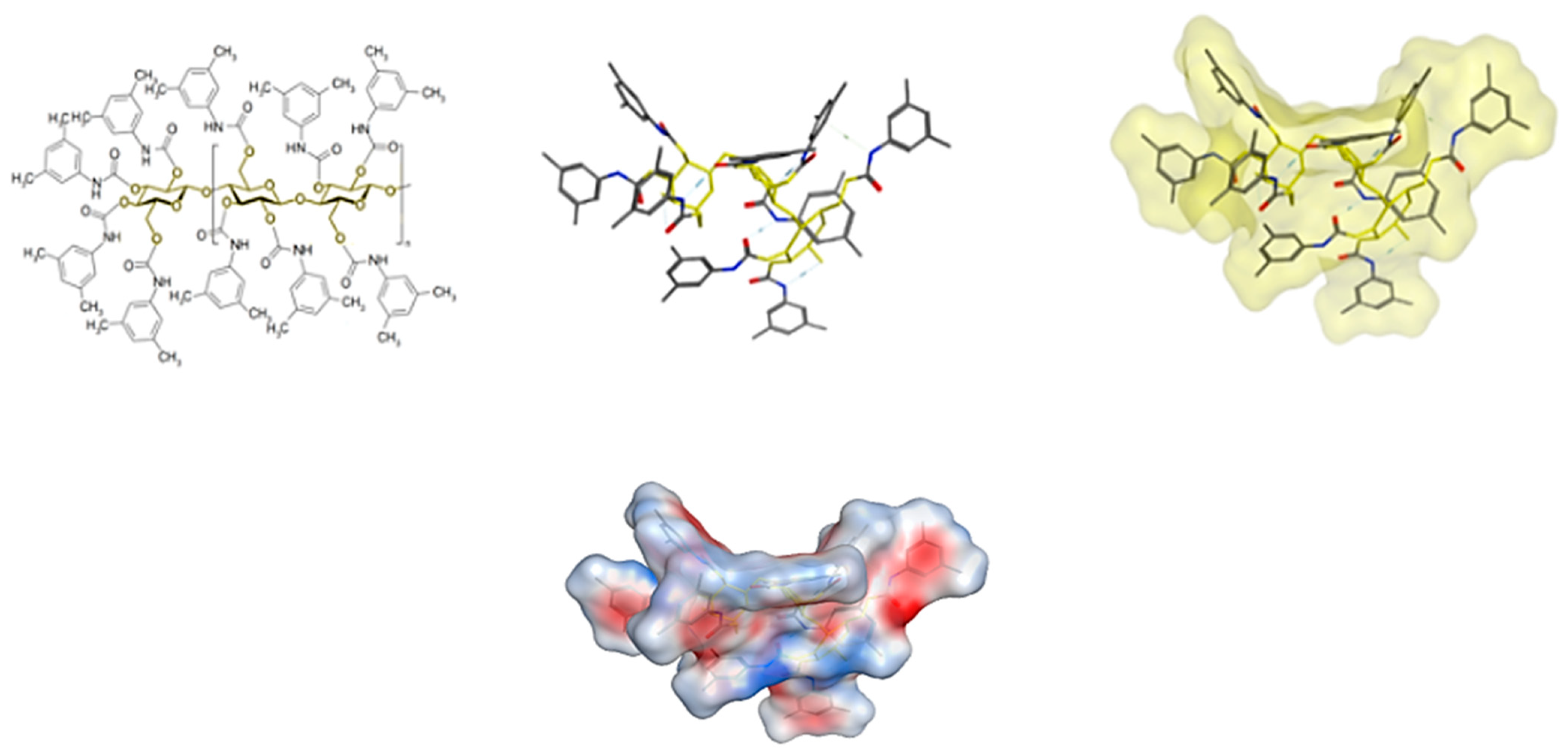
2.4. Computational Study: Investigation of Analyte-CSP Affinities and Identification of Possible Binding Sites and Mechanisms
- -
- H-bonding between the -OH at the chiral center and the carbonyl in CSP
- -
- H-bonding between carbazole-N and the carbonyl group in CSP
- -
- H-bonding between -OH in the substituted phenyl with carbonyl in CSP
- -
- H-π interactions between the analyte’s (substituted) phenyl moiety and the six membered ring in CSP
- -
- H-π bonds between phenyl and carbazole rings and six membered ring in CSP
- -
- π-π interaction between analyte phenyl and the carbonyl group in CSP
- -
- π-π interaction between analyte phenyl and phenyl in CSP
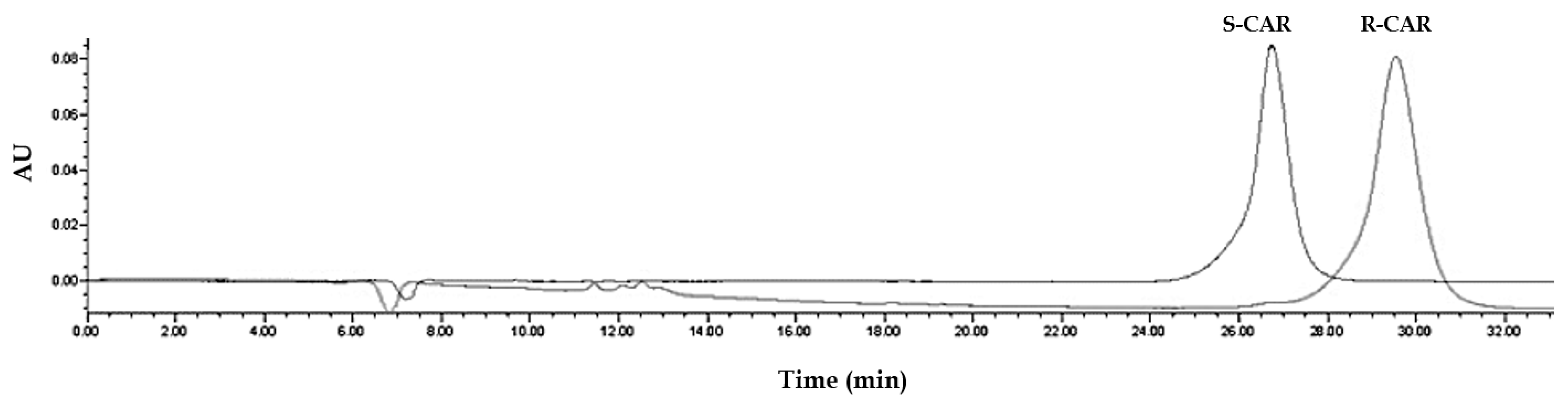
2.5. Experimental Study to Estimate the Enantiomer-Elution Order-No Prediction Possible from Docking Study
2.6. Plasma Concentrations in Patients
3. Discussion and Conclusions
3.1. Method Development and Validation
3.2. Estimation of the Elution Order of Eutomer vs. Distomer and the Respective Metabolite Enantiomers
4. Materials and Methods
4.1. Chemicals
4.2. Equipment and Software
4.3. Stock Solutions, Dilutions for Method Optimization, and Plasma Standard and QC Sample Preparation
4.4. UHPLC-UV Method Development Using a DoE Approach
4.4.1. Screening Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor | Low Level (−1) | High Level (+1) |
|---|---|---|
| Organic modifier composition | Acetonitrile | Acetonitrile: Methanol (1:1, v/v) |
| % of Organic modifier | 60% | 80% |
| Buffer type | 20 mM Ammonium acetate buffer | 20 mM potassium Phosphate buffer |
| Buffer pH | 7.0 | 8.0 |
| Flow rate | 0.5 | 0.7 |
4.4.2. Central Composite Optimization Design
4.4.3. Regression Analysis
4.4.4. Validation of Predictability of Models
4.4.5. Global Solution for Optimum Run Conditions
4.5. Biological Material: Separation of Plasma from Blood
4.6. Sparse Blood Sampling from Patients to Test Assay Applicability
4.7. Plasma Sample Work-Up: Liquid-Liquid Extraction of CAR and Its Major Oxidative Metabolites from Patient Plasma
4.8. Preliminary Method Validation for CAR Enantiomers and the Enantiomers of Its Oxidative Metabolites in Plasma
4.9. Computational Study of the Binding of Analytes to the Chiral Selector
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Poole-Wilson, P.A.; Swedberg, K.; Cleland, J.G.; Di Lenarda, A.; Hanrath, P.; Komajda, M.; Lubsen, J.; Lutiger, B.; Metra, M.; Remme, W.J.; et al. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol or Metoprolol European Trial (COMET): Randomised controlled trial. Lancet 2003, 362, 7–13. [Google Scholar] [CrossRef] [Green Version]
- Morgan, T. Clinical Pharmacokinetics and Pharmacodynamics of Carvedilol. Clin. Pharmacokinet. 1994, 26, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Nichols, A.J.; Sulpizio, A.C.; Ashlon, D.J.; Hiehte, J.P.; Ruffolo, R.R. In vitro pharmacologic profile of the novel beta-adrenoceptor antagonist and vasodilator, carvedilol. Pharmacology 1989, 39, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, W.; Sponer, G.; Strein, K.; Müller-Beckmann, B.; Kling, L.; Böhm, E.; Martin, U.; Borbe, H.O. Pharmacological characteristics of the stereoisomers of carvedilol. Eur. J. Clin. Pharmacol. 1990, 38, 104–107. [Google Scholar] [CrossRef]
- Easson, L.H.; Stedman, E. Studies on the relationship between chemical constitution and physiological action: Molecular dissymmetry and physiological activity. Biochem. J. 1933, 27, 1257. [Google Scholar] [CrossRef]
- Spahn, H.; Henke, W.; Langguth, P.; Schloos, J.; Mutschler, E. Measurement of Carvedilol Enantiomers in Human Plasma and Urine Using S-Naproxen Chloride for Chiral Derivatization. Arch. der Pharm. 1990, 323, 465–469. [Google Scholar] [CrossRef]
- Neugebauer, G.; Akpan, W.; Möllendorff, E.V.; Neubert, P.; Reiff, K. Pharmacokinetics and disposition of carvedilol in humans. J. Cardiovasc. Pharmacol. 1987, 10, S85–S88. [Google Scholar] [CrossRef]
- Takekuma, Y.; Takenaka, T.; Kiyokawa, M.; Yamazaki, K.; Okamoto, H.; Kitabatake, A.; Tsutsui, H.; Sugawara, M. Contribution of polymorphisms in UDP-glucuronosyltransferase and CYP2D6 to the individual variation in disposition of carvedilol. J. Pharm. Pharm. Sci. 2006, 9, 101–112. [Google Scholar]
- Saito, M.; Kawana, J.; Ohno, T.; Hanada, K.; Kaneko, M.; Mihara, K.; Shiomi, M.; Nagayama, M.; Sumiyoshi, T.; Ogata, H. Population pharmacokinetics of R- and S-carvedilol in japanese patients with chronic heart failure. Biol. Pharm. Bull. 2010, 33, 1378–1384. [Google Scholar] [CrossRef] [Green Version]
- Sehrt, D.; Meineke, I.; Tzvetkov, M.; Gültepe, S.; Brockmöller, J. Carvedilol pharmacokinetics and pharmacodynamics in relation to CYP2D6 and ADRB pharmacogenetics. Pharmacogenomics 2011, 12, 783–795. [Google Scholar] [CrossRef]
- Honda, M.; Ogura, Y.; Toyoda, W.; Taguchi, M.; Nozawa, T.; Inoue, H.; Hashimoto, Y. Multiple regression analysis of pharmacogenetic variability of carvedilol disposition in 54 healthy Japanese volunteers. Biol. Pharm. Bull. 2006, 29, 772–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myung, S.W.; Jo, C.H. Gas chromatograph-mass spectrometric method for the determination of carvedilol and its metabolites in human urine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2005, 822, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg EJ, P.W. High-performance Liquid Chromatographic Method for the Simultaneous Determination of the Enantiomers of Carvedilol and its O-desmethyl Metabolite in Human Plasma after Chiral Derivatization. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1989, 493, 105–115. [Google Scholar] [CrossRef]
- Kurz, A. Stereospecific Pharmacokinetics of Carvedilol and its Metabolites 2′-desmethyl carvedilol, 4′-hydroxy Carvedilol, and 5′-hydroxy carvedilol. Drug Metab. Dispos. 1997, 25, 970–977. [Google Scholar]
- Yang, E.; Wang, S.; Kratz, J.; Cyronak, M.J. Stereoselective analysis of carvedilol in human plasma using HPLC/MS/MS after chiral derivatization. J. Pharm. Biomed. Anal. 2004, 36, 609–615. [Google Scholar] [CrossRef]
- Peccinini, R.G.; Ximenes, V.F.; Cesarino, E.J.; Lanchote, V.L. Stereoselective analysis of carvedilol in human plasma and urine using HPLC after chiral derivatization. Biopharm. Drug Dispos. 2008, 29, 280–288. [Google Scholar] [CrossRef]
- Furlong, M.T.; He, B.; Mylott, W.; Zhao, S.; Mariannino, T.; Shen, J.; Stouffer, B. A validated enantioselective LC-MS/MS assay for the simultaneous determination of carvedilol and its pharmacologically active 4′-hydroxyphenyl metabolite in human plasma: Application to a clinical pharmacokinetic study. J. Pharm. Biomed. Anal. 2012, 70, 574–579. [Google Scholar] [CrossRef]
- Clohs, L.; McErlane, K.M. Development of a capillary electrophoresis assay for the determination of carvedilol enantiomers in serum using cyclodextrins. J. Pharm. Biomed. Anal. 2001, 24, 545–554. [Google Scholar] [CrossRef]
- Lamprecht, G.; Gruber, L.; Stoschitzky, K.; Lindner, W. Enantioselective analysis of (R)- and (S)-carvedilol in Human Plasma by High-performance liquid chromatography. Chromatographia 2002, 56, 25–29. [Google Scholar] [CrossRef]
- Saito, M.; Kawana, J.; Ohno, T.; Kaneko, M.; Mihara, K.; Hanada, K.; Sugita, R.; Okada, N.; Oosato, S.; Nagayama, M.; et al. Enantioselective and highly sensitive determination of carvedilol in human plasma and whole blood after administration of the racemate using normal-phase high-performance liquid chromatography. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 843, 73–77. [Google Scholar] [CrossRef]
- Nardotto, G.H.B.; Coelho, E.B.; Marques, M.P.; Lanchote, V.L. Chiral analysis of carvedilol and its metabolites hydroxyphenyl carvedilol and O-desmethyl carvedilol in human plasma by liquid chromatography-tandem mass spectrometry: Application to a clinical pharmacokinetic study. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1015, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Stahl, E.; Mutschler, E.; Baumgartner, U.; Spahn-Langguth, H. Carvedilol Stereopharmacokinetics in Rats: Affinities to Blood Constituents and Tissues. Arch. Pharm. 1993, 326, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Available online: ICH-guideline-m10-bioanalytical-method-validation-step-2b_en.pdf (accessed on 23 June 2020).
- George, N.; Herz, M.; Aboul-Enein, H.Y.; Shihata, L.; Hanafi, R. Surface design of enantiomeric HPLC separation on vancomycin and teicoplanin-based stationary phases, a tool for chiral recognition of model β-blockers. J. Chromatogr. Sci. 2019, 57, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Hanafi, R.S.; Lämmerhofer, M. Response surface methodology for the determination of the design space of enantiomeric separations on cinchona-based zwitterionic chiral stationary phases by high performance liquid chromatography. J. Chromatogr. A 2018, 1534, 55–63. [Google Scholar] [CrossRef]
- Yilmaz, B.; Arslan, S. HPLC/fluorometric detection of carvedilol in real human plasma samples using liquid-liquid extraction. J. Chromatogr. Sci. 2016, 54, 413–418. [Google Scholar] [CrossRef] [Green Version]












| Run Order | % Organic Modifier | Buffer pH | % Acetonitrile in Organic Modifier | Rs | t’R |
|---|---|---|---|---|---|
| 1 | 80 | 7.0 | 80 | 0.77 | 17.6 |
| 2 | 70 | 7.5 | 70 | 0.60 | 45.1 |
| 3 | 80 | 8.0 | 80 | 0.77 | 23.1 |
| 4 | 70 | 7.5 | 70 | 0.63 | 47.1 |
| 5 | 70 | 8.2 | 70 | 0.64 | 68.0 |
| 6 | 70 | 7.5 | 87 | 0.70 | 35.6 |
| 7 | 70 | 6.7 | 70 | 0.46 | 58.8 |
| 8 | 53 | 7.5 | 70 | 0.45 | 175.8 |
| 9 | 60 | 8.0 | 60 | 0.74 | 176.4 |
| 10 | 70 | 7.5 | 70 | 0.60 | 46.3 |
| 11 | 60 | 8.0 | 80 | 0.67 | 92.4 |
| 12 | 60 | 7.0 | 80 | 0.00 | 81.6 |
| 13 | 70 | 7.5 | 70 | 0.64 | 47.3 |
| 14 | 70 | 7.5 | 70 | 0.65 | 47.8 |
| 15 | 87 | 7.5 | 70 | 0.56 | 15.4 |
| 16 | 70 | 7.5 | 53 | 0.66 | 71.9 |
| 17 | 80 | 8.0 | 60 | 0.52 | 26.5 |
| 18 | 60 | 7.0 | 60 | 0.00 | 151.8 |
| 19 | 70 | 7.5 | 70 | 0.60 | 45.6 |
| 20 | 80 | 7.0 | 60 | 0.46 | 24.40 |
| Parameters | CAR | 4′OH-C | 5′OH-C | DMC |
|---|---|---|---|---|
| Linearity range (ng/mL) | 5–100 | 1.25–20 | 1.25–20 | 1.25–20 |
| Regression equation X: Concentration (ng/mL) Y: Peak area ratio | Y = 0.0508x − 0.0029 | Y = 0.1374x − 0.0397 | Y = 0.1500x − 0.0329 | Y = 0.0935x + 0.0705 |
| SD of slope | 1.320 × 10−4 | 7.000 × 10−4 | 2.050 × 10−3 | 2.020 × 10−3 |
| SD of y-intercept | 6.900 × 10−5 | 3.120 × 10−3 | 2.630 × 10−3 | 2.290 × 10−3 |
| R2 | 0.996 | 0.997 | 0.997 | 0.994 |
| SD of R2 | 2.030 × 10−5 | 9.180 × 10−4 | 9.150 × 10−4 | 5.460 × 10−4 |
| LOD | 0.004 | 0.068 | 0.053 | 0.074 |
| LOQ | 0.014 | 0.227 | 0.175 | 0.245 |
| Patient Number | Sampling Time (hr) | Plasma Concentration (ng/mL) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAR | DMC | 4′OHC | 5′OHC | ||||||||||||||
| S-(−) | R-(+) | S/R | rac- | S-(−) | R-(+) | S/R | rac- | S-(−) | R-(+) | S/R | rac- | S-(−) | R-(+) | S/R | rac- | ||
| 1 | 0.5 | 20.28 | 20.70 | 0.98 | 40.98 | - | - | - | - | - | - | - | - | - | - | - | - |
| 2 | 1.0 | 50.01 | 48.39 | 1.00 | 98.40 | 3.27 | 2.40 | 1.36 | 5.67 | - | - | - | - | - | - | - | - |
| 3 | 2.0 | - | - | - | - | 9.92 | 6.06 | 1.51 | 15.98 | 13.74 | 4.51 | 3.05 | 18.25 | 11.48 | 8.20 | 1.40 | 19.68 |
| 4 | 4.0 | - | - | - | - | 8.52 | 4.93 | 1.97 | 13.45 | 16.06 | 4.98 | 3.22 | 21.04 | 14.06 | 9.47 | 1.49 | 23.53 |
| 5 | 9.0 | - | - | - | - | 5.02 | 2.82 | 2.41 | 7.84 | 8.18 | 3.54 | 2.32 | 11.72 | 8.09 | 6.09 | 1.33 | 14.18 |
| 6 | 12.0 | - | - | - | - | 2.73 | 1.52 | 1.97 | 4.25 | 5.29 | 1.96 | 2.70 | 7.25 | 5.28 | 3.78 | 1.37 | 9.06 |
| Molecule | m.w. (g/mol) | cLogP | vdWSA | TPSA | Individual Binding Score (Sc) | R-vs.-S Score-Difference | Capacity Factor k | Separation Factor (α) | Resolution (Rs) |
|---|---|---|---|---|---|---|---|---|---|
| R-CAR | 406.5 | 3.90 | 668 | 75.74 | −20.60 | 4.51 | 4.28 | 1.12 | best resolution 2.4 |
| S-CAR | 406.5 | 3.90 | 661 | 75.74 | −25.11 | 3.72 | |||
| R-5′OHC | 422.5 | 3.40 | 684 | 95.97 | −22.08 | 2.37 | 1.41 | 1.04 | 1.2 |
| S-5′OHC | 422.5 | 3.40 | 683 | 95.97 | −24.45 | 1.31 | |||
| R-DMC | 392.5 | 3.49 | 640 | 86.74 | −18.07 | 2.05 | 2.35 | 1.04 | 1.0 |
| S-DMC | 392.5 | 3.49 | 640 | 86.74 | −20.12 | 2.21 | |||
| R-4′OHC | 422.5 | 3.40 | 684 | 95.97 | −22.71 | 1.07 | 1.22 | 1.07 | worst resolution (critical peaks) 0.8 |
| S-4′OHC | 422.5 | 3.40 | 687 | 95.97 | −23.78 | 1.08 | |||
| R-MET (IS.) | 267.4 | 1.99 | 525 | 50.72 | −15.01 | 0.11 | 0.55 | 1.0 | no resolution0 |
| S-MET (IS) | 267.4 | 1.99 | 523 | 50.72 | −15.12 | 0.55 |
| Factor | Low Level | High Level |
|---|---|---|
| X1: % of organic modifier | 60 | 80 |
| X2: Buffer pH | 7.0 | 8.0 |
| X3: Organic modifier composition: % Acetonitrile of the total organic modifier (remaining is methanol) | 60% | 80% |
| Patient Initials | Number | Gender | Age (yrs) | Weight (kg) | Diagnosis |
|---|---|---|---|---|---|
| HH | 1 | F | 51 | 87 | Ischemic heart failure Diabetes |
| MG | 2 | F | 66 | 91 | Hypertension Gastritis Anemia |
| RS | 3 | M | 55 | 89 | Myocardial infarction Diabetes |
| EI | 4 | M | 47 | 76 | Hypertension Gastritis |
| AS | 5 | F | 53 | 95 | Hypertension Gout |
| KS | 6 | F | 62 | 85 | Hypertension Diabetes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samir, L.; Hanafi, R.; El Deeb, S.; Spahn-Langguth, H. UHPLC Enantiomer Resolution for the ɑ/β-Adrenoceptor Antagonist R/S-Carvedilol and Its Major Active Metabolites on Chiralpak IB N-5. Molecules 2022, 27, 4998. https://doi.org/10.3390/molecules27154998
Samir L, Hanafi R, El Deeb S, Spahn-Langguth H. UHPLC Enantiomer Resolution for the ɑ/β-Adrenoceptor Antagonist R/S-Carvedilol and Its Major Active Metabolites on Chiralpak IB N-5. Molecules. 2022; 27(15):4998. https://doi.org/10.3390/molecules27154998
Chicago/Turabian StyleSamir, Liza, Rasha Hanafi, Sami El Deeb, and Hilde Spahn-Langguth. 2022. "UHPLC Enantiomer Resolution for the ɑ/β-Adrenoceptor Antagonist R/S-Carvedilol and Its Major Active Metabolites on Chiralpak IB N-5" Molecules 27, no. 15: 4998. https://doi.org/10.3390/molecules27154998