
High Throughput Identification of the Potential Antioxidant Peptides in Ophiocordyceps sinensis

Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Data Information
2.2. Gene Annotation and Differential Expression Analysis
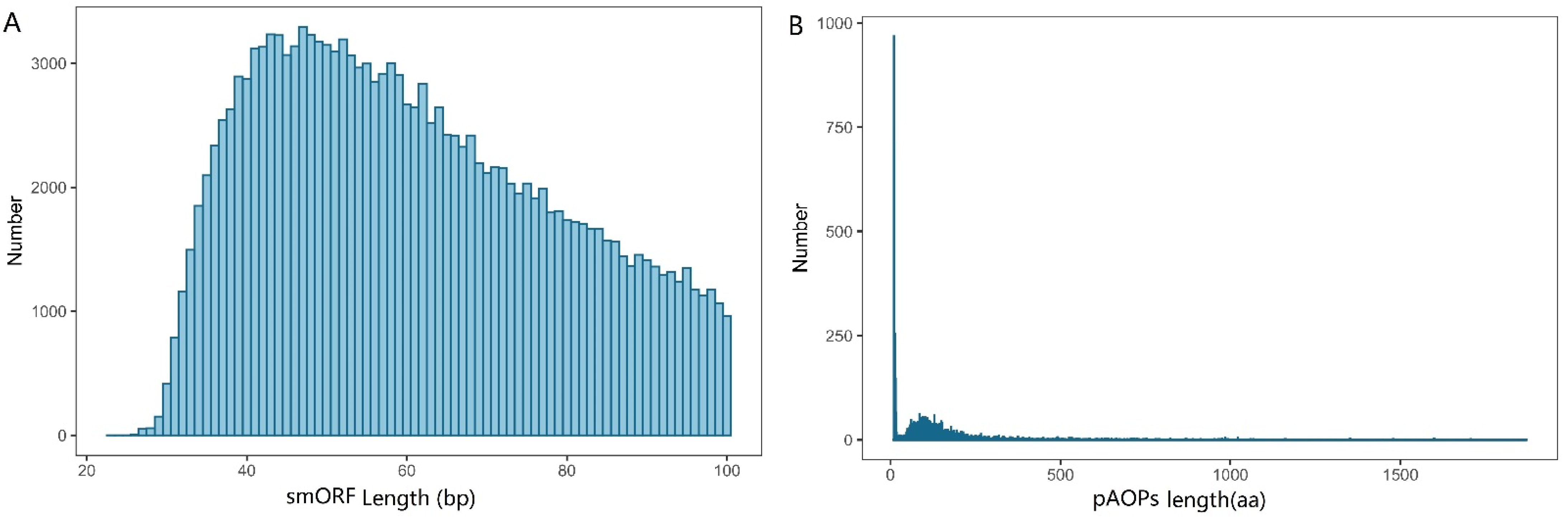
2.3. smORF Identification
2.4. AOP Identification
2.5. Protein Preparation
2.6. Peptide Isobaric Labeling
2.7. HPLC Fractionation and High-Resolution LC–MS/MS Analysis Based on Q Exactive
2.8. Data Processing
2.9. Statistical Analysis
3. Results
3.1. Overview of Transcriptome and Differential Expression Analysis
3.2. AOP Identification and Functional Annotation
3.3. The Differential Expression Analysis of AOPs
3.4. Validation by RT-PCR
3.5. iTRAQ Analysis and pAOPs Validation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| YF | the wild-grown O. sinensis with mature fruit-body |
| MF | the artificial O. sinensis with mature fruit-body |
| IL | the mycoparasite complex |
| ST | the mummified larvae coated with mycelia were designated as the sclerotium |
| RNA seq | RNA-sequencing |
| log2FC | log 2fold-change |
| DEGs | differentially expressed genes |
| DAPs | differentially expressed antioxidant peptides |
| pAOPs | putative antioxidant peptides |
| AMPs | anti-microbial peptides |
| GO | gene ontology analysis |
| TCM | Traditional Chinese of Medicine |
References
- Lo, H.; Hsieh, C.; Lin, F.Y.; Hsu, T.H. A systematic review of the mysterious caterpillar fungus Ophiocordyceps sinensis in Dong-Chong Xia Cao (Dong Chong Xia Căo) and related bioactive ingredients. J. Tradit. Complement. Med. 2013, 3, 16–32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, E.; Wang, C.; Li, Y.; Liu, X. Ophiocordyceps sinensis, the flagship fungus of China: Terminology, life strategy and ecology. Mycology 2012, 3, 2–10. [Google Scholar] [CrossRef]
- Qin, Q.; Zhou, G.L.; Zhang, H.; Meng, Q.; Zhang, J.; Wang, H.; Miao, L.; Li, X. Obstacles and approaches in artificial cultivation of Chinese cordyceps. Mycology 2018, 9, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Huang, Y.; Chen, X.; Mo, M. The Mechanisms of Pharmacological Activities of Ophiocordyceps sinensis Fungi. Phytother. Res. 2016, 30, 1572–1583. [Google Scholar] [CrossRef] [PubMed]
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C.; et al. A Review of the Microbial Production of Bioactive Natural Products and Biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Liu, Q.; Li, W.; Li, Q.; Qian, Z.; Liu, X.; Dong, C. A breakthrough in the artificial cultivation of Chinese cordyceps on a large-scale and its impact on science, the economy, and industry. Crit. Rev. Biotechnol. 2019, 39, 181–191. [Google Scholar] [CrossRef]
- Shrestha, U.B. Asian medicine: A fungus in decline. Nature 2012, 482, 35. [Google Scholar] [CrossRef] [Green Version]
- Xia, E.; Yang, D.R.; Jiang, J.; Zhang, Q.; Liu, Y.; Liu, Y.; Zhang, Y.; Zhang, H.; Shi, C.; Tong, Y.; et al. The caterpillar fungus, Ophiocordyceps sinensis, genome provides insights into highland adaptation of fungal pathogenicity. Sci. Rep. 2017, 7, 1806–1817. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, C.D.; Patsoukis, N.; Papapostolou, I.; Zervoudakis, G. Sclerotial metamorphosis in filamentous fungi is induced by oxidative stress. Integr. Comp. Biol. 2006, 46, 691–712. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, Z.; Dong, H.; Dong, C. Reactive oxygen species induce sclerotial formation in Morchella importuna. Appl. Microbiol. Biotechnol. 2018, 102, 7997–8009. [Google Scholar] [CrossRef]
- Yoshida, Y.; Hasunuma, K. Reactive oxygen species affect photomorphogenesis in Neurosporacrassa. J. Biol. Chem. 2004, 279, 6986–6993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belozerskaya, T.A.; Gessler, N.N.; Isakova, E.P.; Deryabina, Y.I. Neurospora crassa light signal transduction is affected by ROS. J. Signal Transduct. 2012, 2012, 791963. [Google Scholar] [CrossRef] [Green Version]
- Lara-Ortíz, T.; Riveros-Rosas, H.; Aguirre, J. Reactive oxygen species generated by microbial NADPH oxidase NoxA regulate sexual development in Aspergillusnidulans. Mol. Microbiol. 2003, 50, 1241–1255. [Google Scholar] [CrossRef] [Green Version]
- Krijgsheld, P.; Bleichrodt, R.; Van Veluw, G.; Wang, F.; Müller, W.H.; Dijksterhuis, J.; Wösten, H.A.B. Development in Aspergillus. Stud. Mycol. 2013, 74, 1–29. [Google Scholar] [CrossRef]
- Li, X.; Wang, F.; Liu, Q.; Li, Q.P.; Qian, Z.H.; Zhang, X.L.; Li, K.; Li, W.J.; Dong, C.H. Developmental transcriptomics of Chinese cordyceps reveals gene regulatory network and expression profiles of sexual development-related gene. BMC Genom. 2019, 20, 337. [Google Scholar] [CrossRef]
- Tong, X.; Wang, F.; Zhang, H.; Bai, J.; Dong, Q.; Yue, P.; Jiang, X.; Li, X.; Wang, L.; Guo, J. iTRAQ-based comparative proteome analyses of different growth stages revealing the regulatory role of reactive oxygen species in the fruiting body development of Ophiocordyceps sinensis. PeerJ 2021, 9, e10940. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Guo, S.; Liu, H. Antioxidant activity and inhibition of ultraviolet radiation-induced skin damage of Selenium-rich peptide fraction from selenium-rich yeast protein hydrolysate. Bioorg. Chem. 2020, 105, 104431. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 15, 3674–3676. [Google Scholar] [CrossRef] [Green Version]
- Feng, P.; Chen, W.; Lin, H. Identifying Antioxidant Proteins by Using Optimal Dipeptide Compositions. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 186–191. [Google Scholar] [CrossRef]
- Olsen, T.H.; Yesiltas, B.; Marin, F.I.; Pertseva, M.; García-Moreno, P.J.; Gregersen, S.; Overgaard, M.T.; Jacobsen, C.; Lund, O.; Hansen, E.B.; et al. AnOxPePred: Using deep learning for the prediction of antioxidative properties of peptides. Sci. Rep. 2020, 10, 21471. [Google Scholar] [CrossRef]
- Webb-Robertson, B.J.; Cannon, W.R.; Oehmen, C.S.; Shah, A.R.; Gurumoorthi, V.; Lipton, M.S.; Waters, K.M. A support vector machine model for the prediction of proteotypic peptides for accurate mass and time proteomics. Bioinformatics 2010, 26, 1677–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.S.; Wang, L.; Guo, F.; Zou, Q. AOPs-SVM: A Sequence-Based Classifier of Antioxidant Proteins Using a Support Vector Machine. Front. Bioeng. Biotechnol. 2019, 7, 224. [Google Scholar] [CrossRef]
- Bodzon-Kulakowska, A.; Dylag, T.; Drabik, P.S.; Noga, M.; Jarzebinska, J.; Silberring, J. Methods for samples preparation in proteomic research. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 848, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; He, Y.; Lin, Z.; Zi, J.; Yang, H.; Zhang, S.; Lou, X.; Wang, Q.; Li, S.; Liu, S. Reagents for Isobaric Labeling Peptides in Quantitative Proteomics. Anal.Chem. 2018, 90, 12366–12371. [Google Scholar] [CrossRef]
- Petras, D.; Koester, I.; Da Silva, R.; Stephens, B.M.; Haas, A.F.; Nelson, C.E.; Kelly, L.W.; Aluwihare, L.I.; Dorrestein, P.C. High-Resolution Liquid Chromatography Tandem Mass Spectrometry Enables Large Scale Molecular Characterization of Dissolved Organic Matter. Front. Mar. Sci. 2017, 4, 405. [Google Scholar] [CrossRef] [Green Version]
- Rao, X.; Huang, X.; Zhou, Z.; Lin, X. An improvement of the 2ˆ(-delta delta CT) method for quantitative real-time polymerase chain reaction data analysis. Biostat. Bioinform. Biomath. 2013, 3, 71–85. [Google Scholar]
- Zhong, X.; Gu, L.; Wang, H.; Lian, D.; Zheng, Y.; Zhou, S.; Zhou, W.; Gu, J.; Zhang, G.; Liu, X. Profile of Ophiocordyceps sinensis transcriptome and differentially expressed genes in three different mycelia, Sclerotium and fruiting body developmental stages. Fungal Biol. 2018, 122, 943–951. [Google Scholar] [CrossRef]
- Tong, X.; Zhang, H.; Wang, F.; Xue, Z.; Cao, J.; Peng, C.; Guo, J. Comparative transcriptome analysis revealed genes involved in the fruiting body development of Ophiocordyceps sinensis. PeerJ 2020, 8, e8379. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Huff, L.P.; Fujii, M.; Griendling, K.K. Redox regulation of the actin cytoskeleton and its role in the vascular system. Free Radic. Biol. Med. 2017, 109, 84–107. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol. 1997, 82, 291–295. [Google Scholar] [CrossRef]
- Lushchak, V.I. Adaptive response to oxidative stress: Bacteria, fungi, plants and animals. Comp. Biochem. Physiol. C Toxicol. Pharm. 2011, 153, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Gow, N.A.; Erwig, L.P. Novel insights into host-fungal pathogen interactions derived from live-cell imaging. Semin. Immunopathol. 2015, 37, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Zhao, Z.; Zhuo, T.; Fan, X.; Zou, H. The RSc0454-Encoded FAD-Linked Oxidase Is Indispensable for Pathogenicity in Ralstonia solanacearum GMI1000. Mol. Plant Microbe Interact. 2019, 32, 697–707. [Google Scholar] [CrossRef]
- Borneman, A.R.; Hynes, M.J.; Andrianopoulos, A. BAn STE12 homolog from the asexual, dimorphic fungus Penicillium marneffei complements the defect in sexual development of an Aspergillus nidulans steA mutant. Genetics 2001, 157, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Zhu, W.; Cheng, J.; Xie, J.; Jiang, D.; Li, G.; Chen, W.; Fu, Y. Nox Complex signal and MAPK cascade pathway are cross-linked and essential for pathogenicity and conidiation of mycoparasite Coniothyrium minitans. Sci. Rep. 2016, 6, 24325. [Google Scholar] [CrossRef] [Green Version]
- Chong, W.C.; Shastri, M.D.; Eri, R. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Nexus Implicated in Bowel Disease Pathophysiology. Int. J. Mol. Sci. 2017, 18, 771. [Google Scholar] [CrossRef]
- Ha, H.-J.; Subburaj, S.; Kim, Y.-S.; Kim, J.-B.; Kang, S.-Y.; Lee, G.-J. Molecular Characterization and Identification of Calnexin 1 As a Radiation Biomarker from Tradescantia BNL4430. Plants 2020, 9, 387. [Google Scholar] [CrossRef] [Green Version]
- Dietz, K.J. Peroxiredoxins in plants and cyanobacteria. Antioxid. Redox Signal. 2011, 15, 1129–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ismail, T.; Kim, Y.; Lee, H.; Lee, D.S.; Lee, H.S. Interplay Between Mitochondrial Peroxiredoxins and ROS in Cancer Development and Progression. Int. J. Mol. Sci. 2019, 20, 4407. [Google Scholar] [CrossRef] [Green Version]
- Garrigós, V.; Picazo, C.; Matallana, E.; Aranda, A. Wine yeast peroxiredoxin TSA1 plays a role in growth, stress response and trehalose metabolism in biomass propagation. Microorganisms 2020, 8, 1537. [Google Scholar] [CrossRef] [PubMed]
- Li, H.C.; Chen, Q.Z.; Ma, Y.; Zhou, J.F. Imbalanced free radicals and antioxidant defense systems in schizophrenia: A comparative study. J. Zhejiang Univ. Sci. B 2006, 7, 981–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total_Reads | Overall_Align_Ratio |
|---|---|---|
| IL_1 | 23,401,999 | 94.09% |
| IL_2 | 23,360,595 | 94.07% |
| IL_3 | 22,210,722 | 93.87% |
| ST_1 | 25,374,934 | 92.90% |
| ST_2 | 28,656,031 | 93.10% |
| ST_3 | 21,814,291 | 92.56% |
| YF-1 | 22,893,233 | 95.53% |
| YF-2 | 32,326,044 | 95.45% |
| YF-3 | 20,694,375 | 95.09% |
| MF_1 | 23,239,296 | 96.88% |
| MF_2 | 22,212,619 | 91.69% |
| MF_3 | 22,963,667 | 97.68% |
| AOP Gene_Id | AOP Peptide |
|---|---|
| IOZ07G1895.t1.c465-1 | LIGPHSVIGR |
| IOZ07G2719.t1.c853-821 | GRDSTLGEIA |
| IOZ07G6427.t1.1-696 | SYGVLLEDEGVALR |
| MSTRG.10682.1.c1440-1402 | SGCLRPRANHLG |
| The AOPs ID | Swiss-Prot ID | Annotation |
|---|---|---|
| MSTRG.8461 | sp|Q92405|CATB_ASPFU | Catalase B OS |
| MSTRG.3794 | sp|D7UQ40|SOL5_ALTSO | FAD linked oxidase |
| MSTRG.3929 | sp|P38758|TDA3_YEAST | Putative oxidoreductase TDA3 OS |
| MSTRG.3935 | sp|O74628|YQ53_SCHPO | Uncharacterized oxidoreductase C162.03 OS |
| MSTRG.7531 | sp|Q9P7Q7|MAK1_SCHPO | Peroxide stress-activated histidine kinase mak1 OS |
| MSTRG.2357 | sp|Q6Q487|CALX_ASPFU | Calnexin homolog OS |
| IOZ07G3010 | sp|Q10058|YAM3_SCHPO | Putative oxidoreductase C1F5.03c OS |
| IOZ07G1808 | sp|Q8TFM8|THIO_FUSCU | Thioredoxin-like protein OS |
| IOZ07G1895 | sp|Q8J0N3|SODC_ISATE | Superoxide dismutase [Cu–Zn] OS |
| IOZ07G2061 | sp|O43099|PMP20_ASPFU | Putative peroxiredoxin pmp20 OS |
| IOZ07G4162 | sp|P34227|PRX1_YEAST | Mitochondrial peroxiredoxin PRX1 OS |
| IOZ07G6057 | sp|P23710|NDUS3_NEUCR | NADH-ubiquinone oxidoreductase 30.4 kDa subunit, mitochondrial OS |
| IOZ07G6109 | sp|Q9P7Q7|MAK1_SCHPO | Peroxide stress-activated histidine kinase mak1 OS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tong, X.; Guo, J. High Throughput Identification of the Potential Antioxidant Peptides in Ophiocordyceps sinensis. Molecules 2022, 27, 438. https://doi.org/10.3390/molecules27020438
Tong X, Guo J. High Throughput Identification of the Potential Antioxidant Peptides in Ophiocordyceps sinensis. Molecules. 2022; 27(2):438. https://doi.org/10.3390/molecules27020438
Chicago/Turabian StyleTong, Xinxin, and Jinlin Guo. 2022. "High Throughput Identification of the Potential Antioxidant Peptides in Ophiocordyceps sinensis" Molecules 27, no. 2: 438. https://doi.org/10.3390/molecules27020438