
Adsorption Sites on Pd Nanoparticles Unraveled by Machine-Learning Potential with Adaptive Sampling

 , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Methods
2.1. DFT Computation Details
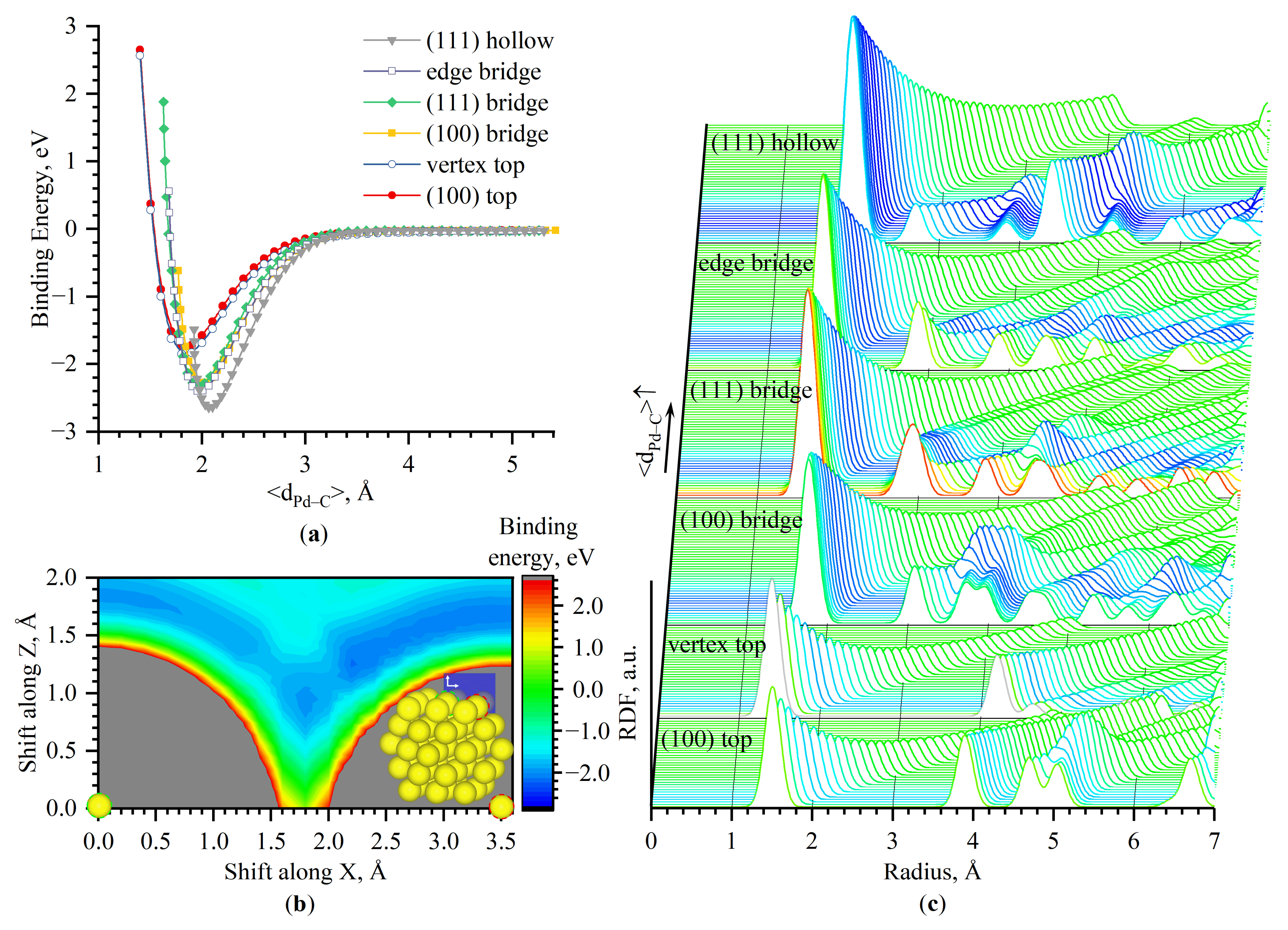
- the central atom of Pd(100)–(100)—top
- between two atoms of Pd(100)–(100)—bridge
- between two atoms of Pd(111)–(111)—bridge
- between three atoms of Pd(111)–(111)—hollow (three-fold)
- between two atoms of the edge between Pd(100) and Pd(111)—edge bridge
- on the single atom of the corner—vertex top
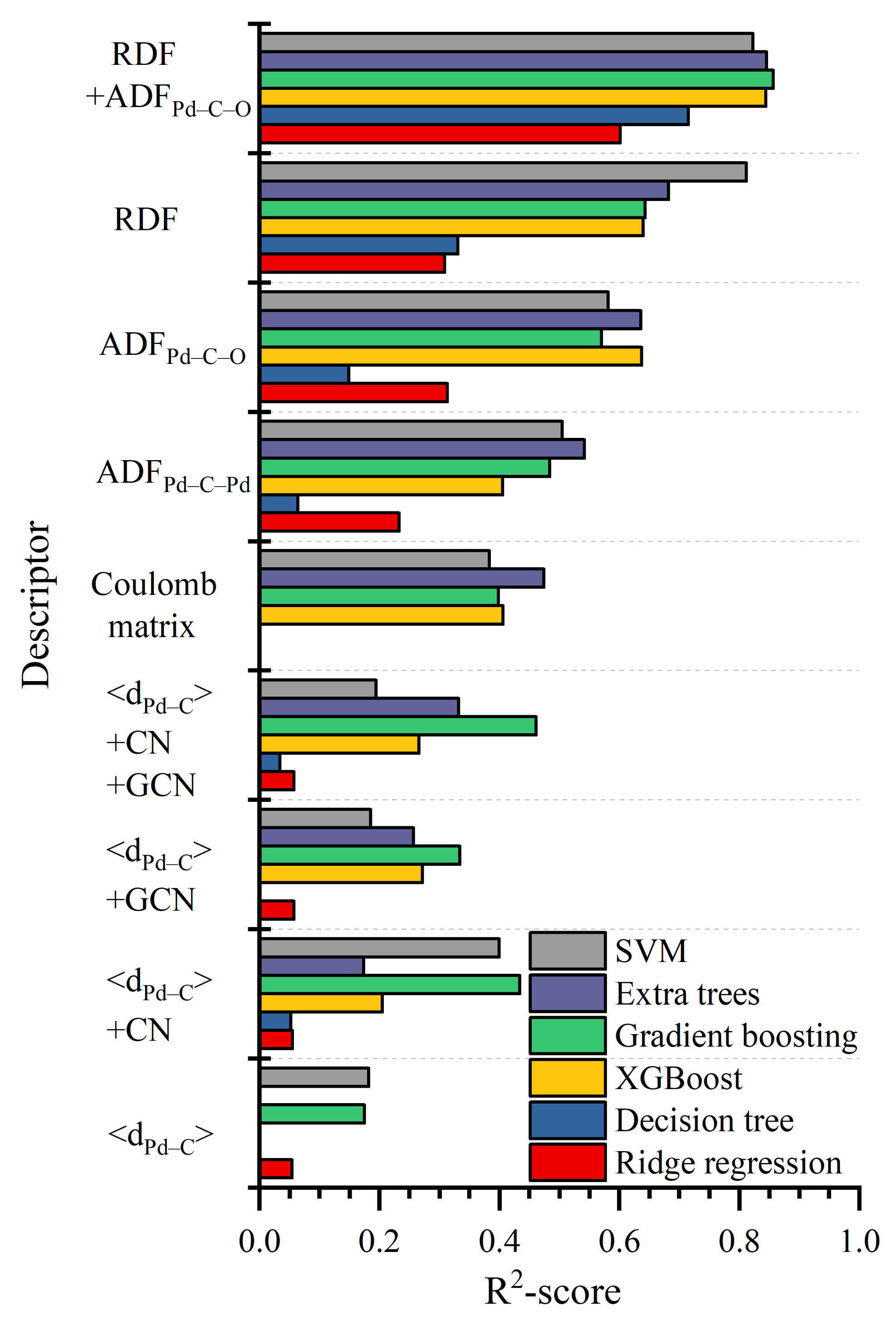
2.2. Descriptors of Structure
- mean distance from carbon to the nearest Pd atoms (<dPd–C>)
- CN of the carbon atom of CO molecule;
- GCN of adsorbing site;
- ADF for Pd–C–Pd and Pd–C–O combinations.
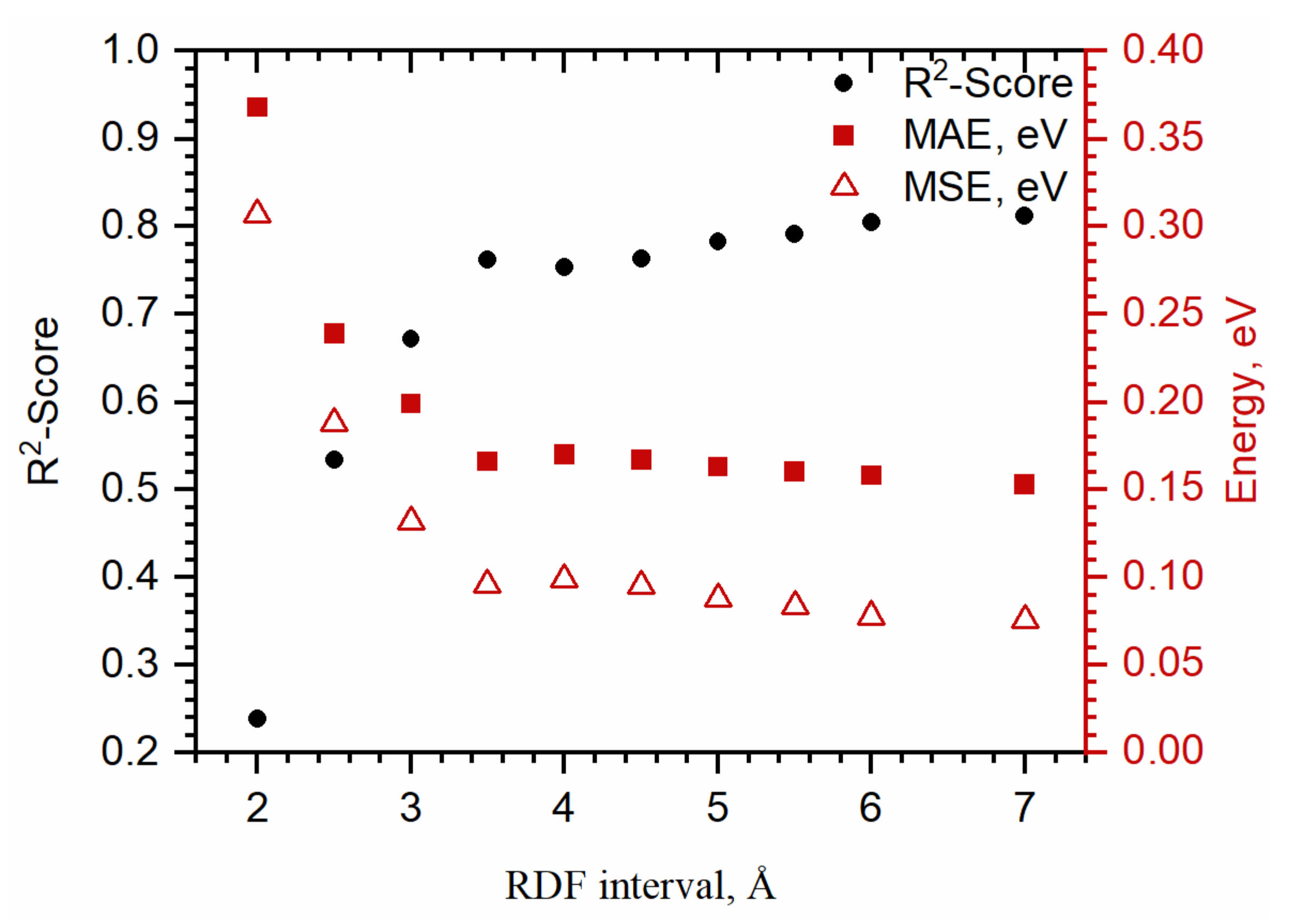
2.3. Training- and Test-Set Preparation
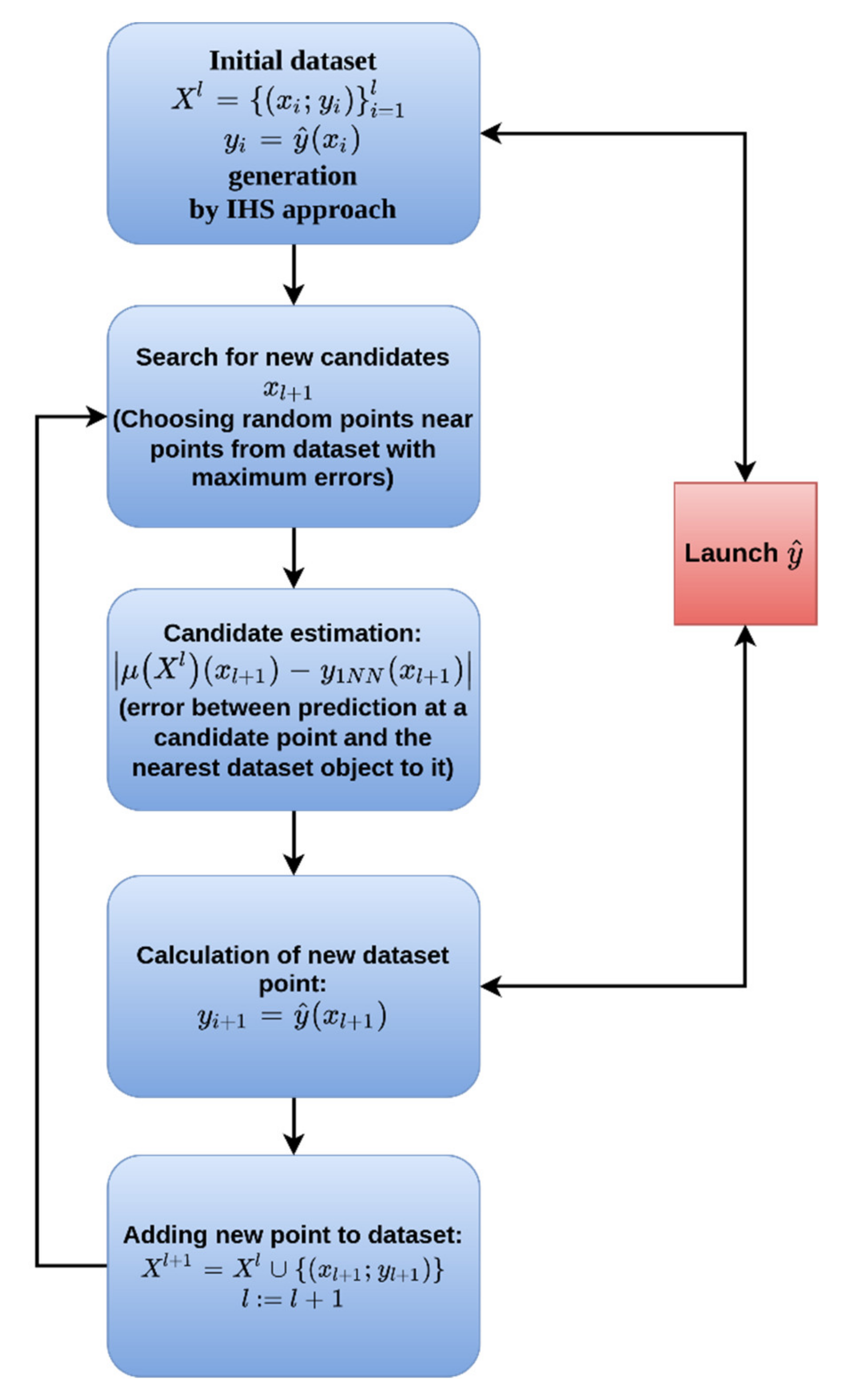
2.4. Adaptive Sampling
2.5. Assessment of Prediction Quality
3. Results and Discussion
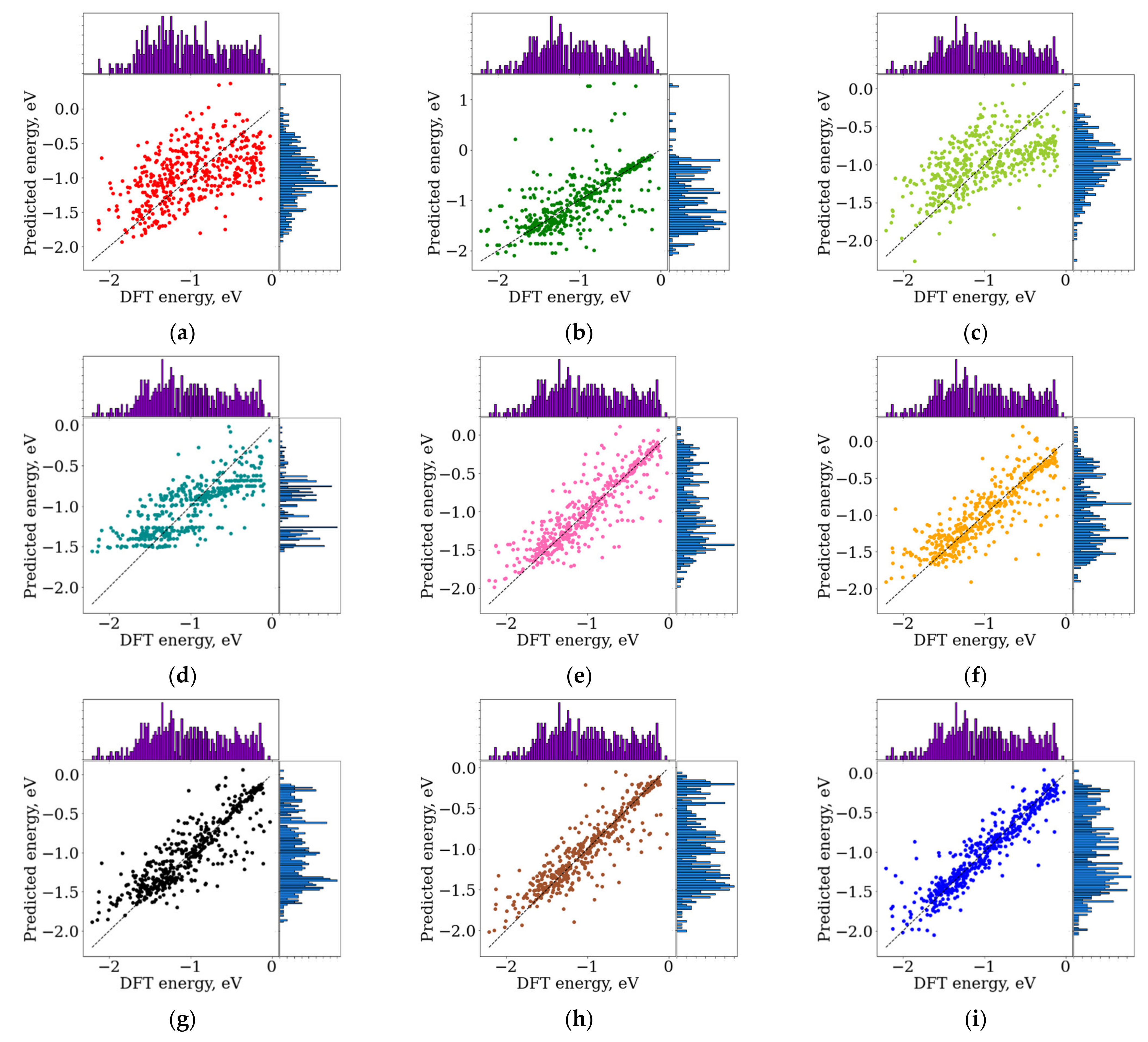
3.1. Prediction Binding Energy Using ASTS
3.2. Comparison between Structural Descriptors
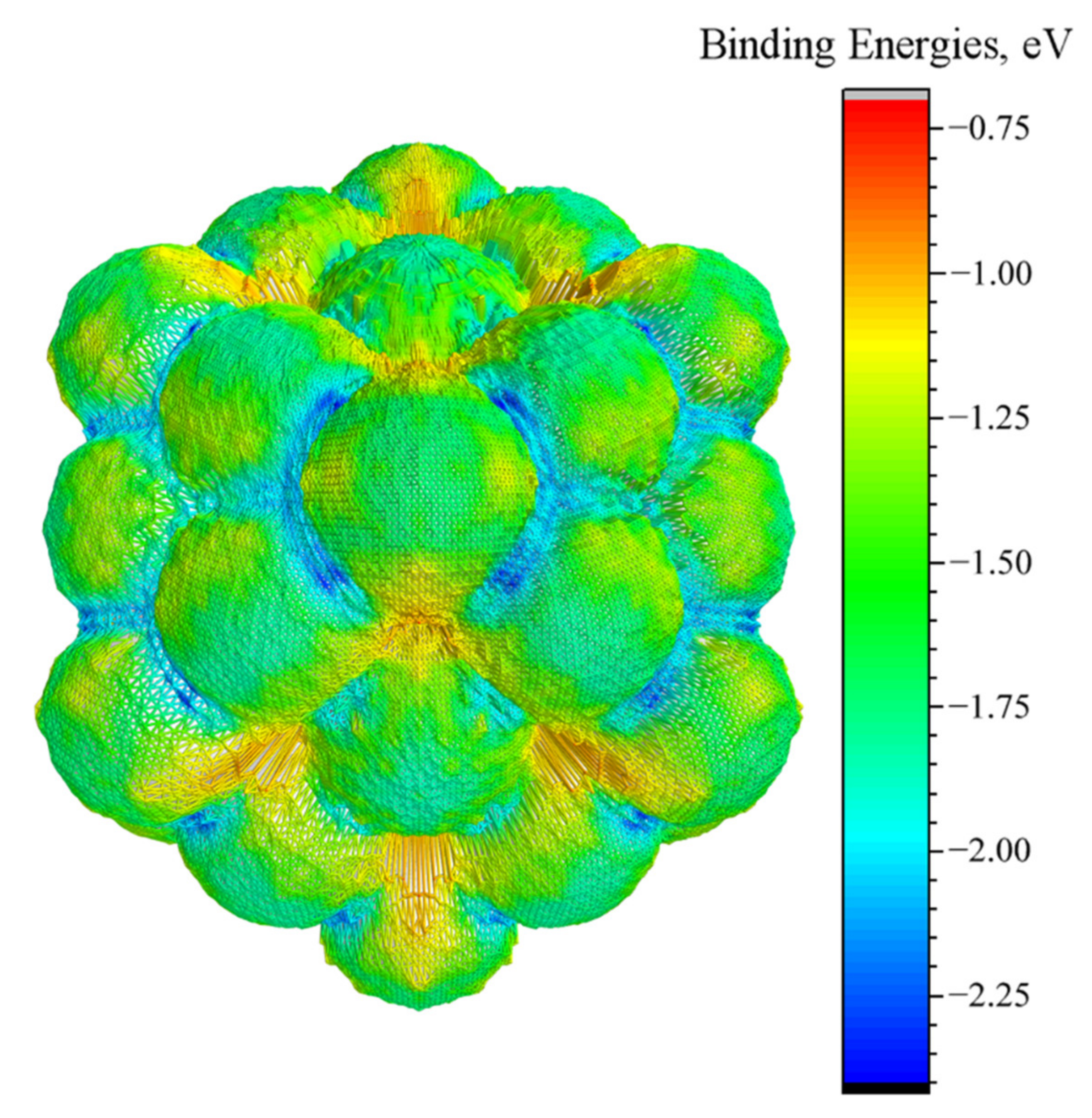
3.3. Energy-Surface Construction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pareek, V.; Bhargava, A.; Gupta, R.; Jain, N.; Panwar, J. Synthesis and applications of noble metal nanoparticles: A review. Adv. Sci. Eng. Med. 2017, 9, 527–544. [Google Scholar] [CrossRef]
- Cuenya, B.R. Synthesis and catalytic properties of metal nanoparticles: Size, shape, support, composition, and oxidation state effects. Thin Solid Film. 2010, 518, 3127–3150. [Google Scholar] [CrossRef]
- Vatti, S.K.; Ramaswamy, K.K.; Balasubramanaian, V. Shape controlled palladium nano particles for hydrogenation of cinnamaldehyde. J. Adv. Nanomat. 2017, 2, 127–132. [Google Scholar] [CrossRef]
- Sun, C.; Cao, Z.; Wang, J.; Lin, L.; Xie, X. Shape and ligand effect of palladium nanocrystals on furan hydrogenation. New J. Chem. 2019, 43, 2567–2574. [Google Scholar] [CrossRef]
- Takeguchi, T.; Manabe, S.; Kikuchi, R.; Eguchi, K.; Kanazawa, T.; Matsumoto, S.; Ueda, W. Determination of dispersion of precious metals on CeO2-containing supports. Appl. Catal. A Gen. 2005, 293, 91–96. [Google Scholar] [CrossRef]
- Shen, M.; Wei, G.; Yang, H.; Wang, J.; Wang, X. Different selections of active sites for CO, C3H6, and C10H22 oxidation on Pd/CeO2 catalysts. Fuel 2013, 103, 869–875. [Google Scholar] [CrossRef]
- Lear, T.; Marshall, R.; Antonio Lopez-Sanchez, J.; Jackson, S.D.; Klapötke, T.M.; Bäumer, M.; Rupprechter, G.; Freund, H.-J.; Lennon, D. The application of infrared spectroscopy to probe the surface morphology of alumina-supported palladium catalysts. J. Chem. Phys. 2005, 123, 174706. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, C.; Zecchina, A.; Groppo, E.; Bordiga, S. Probing the surfaces of heterogeneous catalysts by in situ IR spectroscopy. Chem. Soc. Rev. 2010, 39, 4951–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yudanov, I.V.; Sahnoun, R.; Neyman, K.M.; Rösch, N.; Hoffmann, J.; Schauermann, S.; Johanek, V.; Unterhalt, H.; Rupprechter, G.; Libuda, J. CO adsorption on Pd nanoparticles: Density functional and vibrational spectroscopy studies. J. Phys. Chem. B 2003, 107, 255–264. [Google Scholar] [CrossRef]
- Wang, X.; Wu, G.; Guan, N.; Li, L. Supported Pd catalysts for solvent-free benzyl alcohol selective oxidation: Effects of calcination pretreatments and reconstruction of Pd sites. Appl. Catal. B Environ. 2012, 115, 7–15. [Google Scholar] [CrossRef]
- Aleksandrov, H.A.; Neyman, K.M.; Hadjiivanov, K.I.; Vayssilov, G.N. Can the state of platinum species be unambiguously determined by the stretching frequency of an adsorbed CO probe molecule? Phys. Chem. Chem. Phys. 2016, 18, 22108–22121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichler, A. CO adsorption on Ni (111)—A density functional theory study. Surf. Sci. 2003, 526, 332–340. [Google Scholar] [CrossRef]
- Ouvrard, A.; Wang, J.; Ghalgaoui, A.; Nave, S.; Carrez, S.; Zheng, W.; Dubost, H.; Bourguignon, B. CO Adsorption on Pd (100) Revisited by Sum Frequency Generation: Evidence for Two Adsorption Sites in the Compression Stage. J. Phys. Chem. C 2014, 118, 19688–19700. [Google Scholar] [CrossRef]
- Davis, J.B.; Horswell, S.L.; Piccolo, L.; Johnston, R.L. Computational study of the adsorption of benzene and hydrogen on palladium–iridium nanoalloys. J. Organomet. Chem. 2015, 792, 190–193. [Google Scholar] [CrossRef]
- Zeinalipour-Yazdi, C.D.; Willock, D.J.; Thomas, L.; Wilson, K.; Lee, A.F. CO adsorption over Pd nanoparticles: A general framework for IR simulations on nanoparticles. Surf. Sci. 2016, 646, 210–220. [Google Scholar] [CrossRef]
- Fan, T.-E.; Demiroglu, I.; Hussein, H.A.; Liu, T.-D.; Johnston, R.L. DFT study of the structure, chemical ordering and molecular adsorption of Pd–Ir nanoalloys. Phys. Chem. Chem. Phys. 2017, 19, 27090–27098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankine, C.D.; Madkhali, M.M.; Penfold, T.J. A deep neural network for the rapid prediction of X-ray absorption spectra. J. Phys. Chem. A 2020, 124, 4263–4270. [Google Scholar] [CrossRef] [PubMed]
- Timoshenko, J.; Lu, D.; Lin, Y.; Frenkel, A.I. Supervised machine-learning-based determination of three-dimensional structure of metallic nanoparticles. J. Phys. Chem. Lett. 2017, 8, 5091–5098. [Google Scholar] [CrossRef]
- Timoshenko, J.; Anspoks, A.; Cintins, A.; Kuzmin, A.; Purans, J.; Frenkel, A.I. Neural network approach for characterizing structural transformations by X-ray absorption fine structure spectroscopy. Phys. Rev. Lett. 2018, 120, 225502. [Google Scholar] [CrossRef] [Green Version]
- Tupy, S.A.; Karim, A.M.; Bagia, C.; Deng, W.; Huang, Y.; Vlachos, D.G.; Chen, J.G. Correlating ethylene glycol reforming activity with in situ EXAFS detection of Ni segregation in supported NiPt bimetallic catalysts. ACS Catal. 2012, 2, 2290–2296. [Google Scholar] [CrossRef]
- Lansford, J.L.; Vlachos, D.G. Infrared spectroscopy data-and physics-driven machine learning for characterizing surface microstructure of complex materials. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Calle-Vallejo, F.; Martínez, J.I.; García-Lastra, J.M.; Sautet, P.; Loffreda, D. Fast prediction of adsorption properties for platinum nanocatalysts with generalized coordination numbers. Angew. Chem. Int. Ed. 2014, 53, 8316–8319. [Google Scholar] [CrossRef]
- Gasper, R.; Shi, H.; Ramasubramaniam, A. Adsorption of CO on low-energy, low-symmetry Pt nanoparticles: Energy decomposition analysis and prediction via machine-learning models. J. Phys. Chem. C 2017, 121, 5612–5619. [Google Scholar] [CrossRef]
- Praveen, C.; Comas-Vives, A. Design of an Accurate Machine Learning Algorithm to Predict the Binding Energies of Several Adsorbates on Multiple Sites of Metal Surfaces. ChemCatChem 2020, 12, 4611–4617. [Google Scholar] [CrossRef]
- Beachkofski, B.; Grandhi, R. Improved distributed hypercube sampling. In Proceedings of the 43rd AIAA/ASME/ASCE/AHS/ASC Structures, Structural Dynamics, and Materials Conference, Denver, CO, USA, 22–25 April 2002; p. 1274. [Google Scholar] [CrossRef]
- Hruska, E.; Abella, J.R.; Nüske, F.; Kavraki, L.E.; Clementi, C. Quantitative comparison of adaptive sampling methods for protein dynamics. J. Chem. Phys. 2018, 149, 244119. [Google Scholar] [CrossRef] [PubMed]
- Pérez, A.; Herrera-Nieto, P.; Doerr, S.; De Fabritiis, G. AdaptiveBandit: A multi-armed bandit framework for adaptive sampling in molecular simulations. J. Chem. Theory Comput. 2020, 16, 4685–4693. [Google Scholar] [CrossRef] [PubMed]
- Lookman, T.; Balachandran, P.V.; Xue, D.; Yuan, R. Active learning in materials science with emphasis on adaptive sampling using uncertainties for targeted design. Npj Comput. Mater. 2019, 5, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Gastegger, M.; Behler, J.; Marquetand, P. Machine learning molecular dynamics for the simulation of infrared spectra. Chem. Sci. 2017, 8, 6924–6935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feibelman, P.J.; Hammer, B.; Nørskov, J.K.; Wagner, F.; Scheffler, M.; Stumpf, R.; Watwe, R.; Dumesic, J. The CO/Pt (111) Puzzle. J. Phys. Chem. B 2001, 105, 4018–4025. [Google Scholar] [CrossRef]
- Mason, S.E.; Grinberg, I.; Rappe, A.M. First-principles extrapolation method for accurate CO adsorption energies on metal surfaces. Phys. Rev. B 2004, 69, 161401. [Google Scholar] [CrossRef] [Green Version]
- Mason, S.E.; Grinberg, I.; Rappe, A.M. Adsorbate–Adsorbate Interactions and Chemisorption at Different Coverages Studied by Accurate ab initio Calculations: CO on Transition Metal Surfaces. J. Phys. Chem. B 2006, 110, 3816–3822. [Google Scholar] [CrossRef]
- Hammer, B.; Hansen, L.B.; Nørskov, J.K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerhof functionals. Phys. Rev. B 1999, 59, 7413. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdew, J.; Burke, K.; Ernzerhof, M. Perdew, burke, and ernzerhof reply. Phys. Rev. Lett. 1998, 80, 891. [Google Scholar] [CrossRef]
- Papanikolaou, K.G.; Darby, M.T.; Stamatakis, M. CO-induced aggregation and segregation of highly dilute alloys: A density functional theory study. J. Phys. Chem. C 2019, 123, 9128–9138. [Google Scholar] [CrossRef] [Green Version]
- Github.com. Available online: https://github.com/bjmorgan/vasppy (accessed on 14 November 2021).
- Jain, A.; Hautier, G.; Moore, C.J.; Ong, S.P.; Fischer, C.C.; Mueller, T.; Persson, K.A.; Ceder, G. A high-throughput infrastructure for density functional theory calculations. Comput. Mater. Sci. 2011, 50, 2295–2310. [Google Scholar] [CrossRef]
- Github.com. Available online: https://github.com/materialsproject/pymatgen (accessed on 26 December 2021).
- Muthukrishnan, R.; Rohini, R. LASSO: A feature selection technique in predictive modeling for machine learning. In Proceedings of the 2016 IEEE International Conference on Advances in Computer Applications (ICACA), Coimbatore, India, 24–24 October 2016; pp. 18–20. [Google Scholar] [CrossRef]
- Dietterich, T.G.; Kong, E.B. Machine Learning Bias, Statistical Bias, and Statistical Variance of Decision Tree Algorithms; Technical Report; Department of Computer Science, Oregon State University: Corvallis, OR, USA, 1995; pp. 1–13. Available online: http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.38.2702 (accessed on 5 January 2022).
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- Bennett, K.P.; Campbell, C. Support vector machines: Hype or hallelujah? ACM SIGKDD Explor. Newsl. 2000, 2, 1–13. [Google Scholar] [CrossRef]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobotics 2013, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Geurts, P.; Ernst, D.; Wehenkel, L. Extremely Randomized Trees. Mach. Learn. 2006, 63, 3–42. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD’16), San Francisco, CA, USA, 13–17 August 2016; pp. 785–794.
- Freund, Y.; Schapire, R.E. A decision-theoretic generalization of on-line learning and an application to boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef] [Green Version]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Scikit-learn Machine Learning in Python. Available online: https://scikit-learn.org/ (accessed on 26 December 2021).
- Groppo, E.; Bertarione, S.; Rotunno, F.; Agostini, G.; Scarano, D.; Pellegrini, R.; Leofanti, G.; Zecchina, A.; Lamberti, C. Role of the support in determining the vibrational properties of carbonyls formed on Pd supported on SiO2–Al2O3, Al2O3, and MgO. J. Phys. Chem. C 2007, 111, 7021–7028. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Github.com. Available online: https://github.com/nlesc-nano/auto-FOX (accessed on 5 January 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ML Algorithm | MAE, eV | MSE, eV | R2-Score |
|---|---|---|---|
| Ridge regression | 0.40 | 0.28 | 0.31 |
| Decision tree | 0.30 | 0.27 | 0.33 |
| Lasso | 0.39 | 0.26 | 0.36 |
| AdaBoost | 0.29 | 0.16 | 0.60 |
| XGBoost | 0.20 | 0.15 | 0.64 |
| Gradient boosting | 0.22 | 0.14 | 0.64 |
| Random forest | 0.22 | 0.14 | 0.65 |
| Extra trees | 0.19 | 0.13 | 0.68 |
| SVM | 0.15 | 0.08 | 0.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tereshchenko, A.; Pashkov, D.; Guda, A.; Guda, S.; Rusalev, Y.; Soldatov, A. Adsorption Sites on Pd Nanoparticles Unraveled by Machine-Learning Potential with Adaptive Sampling. Molecules 2022, 27, 357. https://doi.org/10.3390/molecules27020357
Tereshchenko A, Pashkov D, Guda A, Guda S, Rusalev Y, Soldatov A. Adsorption Sites on Pd Nanoparticles Unraveled by Machine-Learning Potential with Adaptive Sampling. Molecules. 2022; 27(2):357. https://doi.org/10.3390/molecules27020357
Chicago/Turabian StyleTereshchenko, Andrei, Danil Pashkov, Alexander Guda, Sergey Guda, Yury Rusalev, and Alexander Soldatov. 2022. "Adsorption Sites on Pd Nanoparticles Unraveled by Machine-Learning Potential with Adaptive Sampling" Molecules 27, no. 2: 357. https://doi.org/10.3390/molecules27020357