4. Materials and Methods
General Methods. All commercially available starting materials and solvents were of reagent grade and used without further purification. Chemical reactions were monitored with thin-layer chromatography using precoated silica gel 60 (0.25 mm thickness) plates. Flash column chromatography was performed on silica gel 60 (SDS 0.040–0.063 mm).
1H NMR spectra were recorded at 298 K in CDCl
3 using the residual signals from CHCl
3 (
1H: = 7.26 ppm) as internal standard.
1H peak assignments were made by first order analysis of the spectra, supported by standard
1H-
1H correlation spectroscopy (COSY) (see
Supplementary Materials).
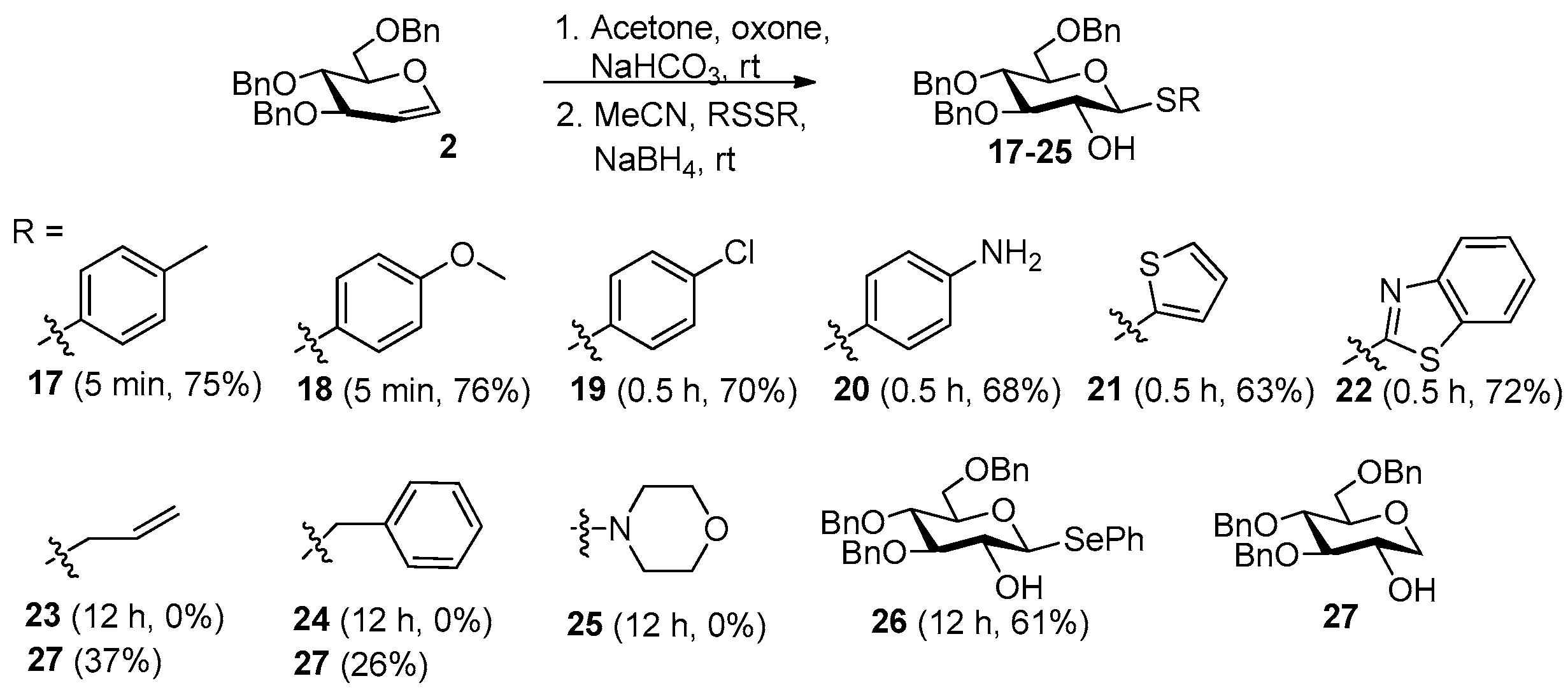
General process A for synthesis of 2-OH 1-thioaryl glycosides from glycals.Step 1. To a cooled (0 °C) solution of a per-protected glycal (1 mmol) in DCM (4 mL) were added acetone (0.4 mL) and saturated aqueous NaHCO3 (7 mL). The mixture was stirred vigorously, and a solution of oxone (2 mmol) in H2O (2.5 mL) was added dropwise over 10 min. The mixture was stirred vigorously at 0 °C for 30 min and then at rt until TLC indicated consumption of the starting material. The organic phase was separated, and the aqueous phase was extracted with DCM (2 × 10 mL). The combined organic phases were dried (MgSO4) and concentrated in vacuo to obtain the crude 1,2-anhydro sugar.
Step 2. To a mixture of phenyl disulfide (or phenyl diselenide) (0.7 mmol) and NaBH4 (53 mg, 1.4 mmol) was added acetonitrile (5 mL). The mixture was stirred at rt for 30 min to 2 h until TLC indicated full conversion of the phenyl disulfide (or phenyl diselenide). The mixture was then added to the crude α-1,2-anhydro sugars. The reaction was stirred at rt for 5–60 min until TLC indicated full conversion of the starting material. The mixture was diluted with DCM and washed with water. The aqueous phase was re-extracted with DCM, and collected organic phases were dried and evaporated under vacuum. The residue was purified by silica gel flash chromatography.
General process B for one-pot synthesis of thioglycoside donors containing a “NGP” group at the 2-position.
Step 1. Same as step 1 in general process A.
Step 2. To a mixture of phenyl disulfide (145 mg, 0.7 mmol) and NaBH4 (53 mg, 1.4 mmol) was added acetonitrile (5 mL). The mixture was stirred at rt for 30 min to 2 h until TLC indicated full conversion of the phenyl disulfide. The mixture was then added to the crude α-1,2-anhydro sugars. The reaction was stirred at rt for 5–60 min until TLC indicated full conversion of the starting material. The reaction mixture was then cooled to 0 °C, followed the slow addition of sodium hydride (6.0 mmol, 6 equiv, 60% oil dispersion), and allowed to stir at 0 °C for 10 min. After that, alkylation/acylation reagents (2–3 equiv) were added to the reaction mixture. The reaction mixture was allowed to warm to rt and then stirred for 1–4 h. Upon completion, the reaction was quenched by adding crushed ice (10 g), stirred until cessation of H2 evolution, and then extracted with ethyl acetate (3 × 80 mL). The combined organic phase was washed with water (3 × 40 mL), separated, dried with MgSO4, and evaporated in vacuo. The residue was purified by column chromatography.
General process C for typical NIS/TfOH-promoted glycosylation procedure. A mixture of a glycosyl donor (0.13 mmol), a glycosyl acceptor (0.10 mmol), and freshly activated molecular sieves (4 Å, 200 mg) in CH2Cl2 (1.6 mL) was stirred under an atmosphere of argon for 1 h. After NIS (0.26 mmol) and TfOH (0.013 mmol) were added at −25 °C, the reaction mixture was allowed to warm to rt over 1 h and then was quenched with TEA and stirred for 30 min. The mixture was then diluted with CH2Cl2, the solid was filtered-off, and the residue was washed with CH2Cl2. After the combined filtrate (30 mL) was washed with water (4 × 10 mL), the organic phase was separated, dried with MgSO4,and concentrated in vacuo. The residue was purified by silica gel flash chromatography.
Phenyl 3,
4,
6-tri-O-benzyl-1-thio-β-D-glucopyranoside (1) [
20]. Following general process A, starting from
2 (100 mg, 0.24 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded
1 as a white solid (94 mg, 72%). Rf = 0.43 (petroleum ether/ethyl acetate 4:1);
1H NMR (600 MHz, chloroform-
d) δ 7.54–7.64 (m, 2H), 7.42–7.25 (m, 16H), 7.24–7.19 (m, 2H), 4.94 (d,
J = 11.2 Hz, 1H), 4.90–4.81 (m, 2H), 4.67–4.55 (m, 3H), 4.53 (d,
J = 9.6 Hz, 1H), 3.83 (dd,
J = 11.0, 2.0 Hz, 1H), 3.77 (dd,
J = 11.0, 4.5 Hz, 1H), 3.67–3.59 (m, 2H), 3.58–3.44 (m, 2H), 2.43 (s, 1H) ppm.
Phenyl 3,4-di-O-benzyl-6-O-tert-butyl-dimethylsily-1-thio-β-D-glucopyranoside (4). Following general process A, starting from 4a (50 mg, 0.114 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 4 as a colorless oil (40 mg, 64%). Rf = 0.41 (petroleum ether/ethyl acetate 8:1); 1H NMR (400 MHz, chloroform-d) δ 7.51–7.43 (m, 2H), 7.31–7.17 (m, 13H), 4.85–4.73 (m, 3H), 4.60 (d, J = 10.8 Hz, 1H), 4.39 (d, J = 9.6 Hz, 1H), 3.86–3.74 (m, 2H), 3.57–3.45 (m, 2H), 3.41–3.32 (m, 1H), 3.28 (ddd, J = 9.2, 3.6, 1.8 Hz, 1H), 2.31 (d, J = 2.1 Hz, 1H), 0.83 (s, 9H), 0.01 (s, 6H) ppm. 13C NMR (100 MHz, chloroform-d) δ 138.49, 138.32, 133.03, 131.69, 128.92, 128.54, 128.47, 128.09, 128.04, 127.95, 127.84, 127.81, 87.89, 85.98, 80.44, 75.43, 75.07, 72.45, 62.14, 25.93, 18.31, −5.11, −5.34 ppm. [α]20D = −20.3 (c 0.32, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5S2Na+, 589.2420; found, 589.2379.
Phenyl 3,4-di-O-benzyl-6-O-acetyl-1-thio-β-D-glucopyranoside (5). Following general process A, starting from 5a (100 mg, 0.271 mmol), after 0.5 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 5 as colorless syrup (93.6 mg, 70%). Rf = 0.58 (petroleum ether/ethyl acetate 4:1); 1H NMR (400 MHz, chloroform-d) δ 7.61–7.50 (m, 2H), 7.43–7.26 (m, 13H), 4.98 (d, J = 11.1 Hz, 1H), 4.93–4.84 (m, 2H), 4.60 (d, J = 10.9 Hz, 1H), 4.52 (d, J = 9.7 Hz, 1H), 4.43 (dd, J = 11.9, 2.1 Hz, 1H), 4.23 (dd, J = 11.9, 5.2 Hz, 1H), 3.70–3.56 (m, 2H), 3.55–3.45 (m, 2H), 2.50 (d, J = 2.2 Hz, 1H), 2.08 (s, 3H) ppm. 13C NMR (100 MHz, chloroform-d) δ 170.67, 138.31, 137.64, 133.04, 131.59, 128.95, 128.57, 128.53, 128.24, 128.10, 128.04, 127.91, 127.00, 88.03, 85.91, 77.19, 75.41, 75.13, 72.72, 66.32, 63.18, 20.86 ppm. [α]20D = −21.6 (c 0.25, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C28H30O6SNa+, 517.1661; found, 517.1640.
Phenyl 3,4-di-O-benzyl-6-O-benzoyl-1-thio-β-D-glucopyranoside (6). Following general process A, starting from 6a (100 mg, 0.232 mmol), after 0.5 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 6 as a colorless oil (81.5 mg, 63%). Rf = 0.36 (petroleum ether/ethyl acetate 6:1); 1H NMR (400 MHz, chloroform-d) δ 8.07–7.93 (m, 2H), 7.60 (t, J = 7.4 Hz, 1H), 7.54–7.05 (m, 17H), 4.96 (d, J = 11.0 Hz, 1H), 4.90–4.80 (m, 2H), 4.69 (dd, J = 12.0, 2.2 Hz, 1H), 4.61 (d, J = 10.8 Hz, 1H), 4.52 (d, J = 9.7 Hz, 1H), 4.44 (dd, J = 11.9, 4.7 Hz, 1H), 3.74–3.63 (m, 2H), 3.59 (t, J = 9.2 Hz, 1H), 3.53–3.44 (m, 1H), 2.46 (d, J = 2.2 Hz, 1H) ppm. 13C NMR (100 MHz, chloroform-d) δ 166.10, 138.25, 137.56, 133.34, 133.16, 131.07, 129.93, 129.77, 128.91, 128.60, 128.54, 128.42, 128.26, 128.16, 128.05, 127.98, 87.78, 85.93, 77.25, 77.01, 75.55, 75.25, 72.55, 63.37 ppm. [α]20D = −31.3 (c 0.15, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5SNa+, 579.1817; found, 579.1803.
Phenyl 3,4,6-tri-O-benzoyl-1-thio-β-D-glucopyranoside (7). Following general process A, starting from 7a (100 mg, 0.218 mmol), after 20 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 7 as colorless syrup (81.6 mg, 64%). Rf = 0.43 (petroleum ether/ethyl acetate 4:1); 1H NMR (400 MHz, chloroform-d) δ 8.07–7.90 (m, 6H), 7.64–7.29 (m, 12H), 7.23–7.18 (m, 2H), 5.66–5.51 (m, 2H), 4.80 (d, J = 9.7 Hz, 1H), 4.67 (dd, J = 12.2, 2.8 Hz, 1H), 4.47 (dd, J = 12.2, 5.8 Hz, 1H), 4.13 (ddd, J = 9.7, 5.7, 2.8 Hz, 1H), 3.77 (t, J = 9.3 Hz, 1H), 2.92–2.88 (m, 1H) ppm. 13C NMR (100 MHz, chloroform-d) δ 166.65, 166.06, 165.35, 133.51, 133.45, 133.41, 133.18, 129.91, 129.86, 129.82, 129.80, 129.67, 129.06, 129.01, 128.73, 128.53, 128.45, 128.41, 128.38, 88.30, 76.52, 76.20, 70.93, 68.93, 63.16 ppm. [α]20D = −20.7 (c 0.058, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C33H28O8SNa+, 607.1367; found, 607.1403.
Phenyl 3,4,6-tri-O-acetyl-1-thio-β-D-glucopyranoside (8). Following general process A, starting from 8a (100 mg, 0.367 mmol), after 15 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 8 as colorless syrup (73.2 mg, 50%). Rf = 0.44 (petroleum ether/ethyl acetate 2:1); 1H NMR (400 MHz, chloroform-d) δ 7.59–7.53 (m, 2H), 7.39–7.28 (m, 3H), 5.13 (t, J = 9.3 Hz, 1H), 4.98 (t, J = 9.8 Hz, 1H), 4.57 (d, J = 9.7 Hz, 1H), 4.25–4.12 (m, 2H), 3.73 (ddd, J = 10.1, 5.0, 2.5 Hz, 1H), 3.50 (td, J = 9.4, 2.8 Hz, 1H), 2.53 (d, J = 2.9 Hz, 1H), 2.09 (s, 3H), 2.07 (s, 3H), 2.03 (s, 3H) ppm. 13C NMR (100 MHz, chloroform-d) δ 170.77, 170.61, 133.59, 130.60, 129.10, 128.72, 88.08, 75.89, 75.77, 70.31, 68.12, 62.24, 20.80, 20.76, 20.62 ppm. [α]20D = −70.0 (c 0.05, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5SNa+, 421.0933; found, 421.0950.
Phenyl 3,4,6-tri-O-ethyl-1-thio-β-D-glucopyranoside (9). Following general process A, starting from 9a (100 mg, 0.435 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 9 as a white solid (99.7 mg, 65%): mp 83.3–84.5 °C; Rf = 0.31 (petroleum ether/ethyl acetate 8:1); 1H NMR (400 MHz, chloroform-d) δ 7.62–7.53 (m, 2H), 7.34–7.26 (m, 3H), 4.49 (d, J = 9.4 Hz, 1H), 3.96–3.77 (m, 3H), 3.73 (dd, J = 11.0, 2.0 Hz, 1H), 3.70–3.48 (m, 4H), 4.44–4.21 (m, 4H), 2.49 (d, J = 2.1 Hz, 1H), 1.27–1.17 (m, 9H) ppm. 13C NMR (100 MHz, chloroform-d) δ 132.73, 128.88, 127.92, 88.04, 85.83, 79.64, 77.58, 72.25, 69.43, 68.73, 68.27, 66.97, 15.79, 15.72, 15.25 ppm. [α]20D = −75.7 (c 0.14, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5S2Na+, 379.1555; found, 379.1558.
Phenyl 3,4,6-tri-O-tert-butyl-dimethylsily-1-thio-β-D-glucopyranoside (10). Following general process A, starting from 10a (100 mg, 0.205 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 10 as a colorless oil (88 mg, 70%); Rf = 0.55 (petroleum ether/ethyl acetate 50:1); 1H NMR (400 MHz, chloroform-d) δ 7.56–7.49 (m, 2H), 7.32–7.19 (m, 3H), 4.59 (d, J = 8.9 Hz, 1H), 3.92 (dd, J = 11.3, 1.9 Hz, 1H), 3.74 (dd, J = 11.3, 5.6 Hz, 1H), 3.58–3.49 (m, 1H), 3.49–3.40 (m, 2H), 3.29 (ddd, J = 9.2, 5.7, 1.9 Hz, 1H), 2.17 (d, J = 2.7 Hz, 1H), 0.97–0.88 (m, 27H), 0.26–0.03 (m, 18H) ppm. 13C NMR (100 MHz, chloroform-d) δ 135.53, 130.74, 128.75, 126.78, 89.09, 81.04, 80.17, 74.40, 70.87, 62.82, 26.14, 25.96, 18.43, 18.24, −3.67, −3.85, −4.08, −4.87, −5.09, −5.33 ppm. [α]20D = −63.8 (c 0.16, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C30H58O5Si3SNa+, 637.3210; found, 637.3174.
Phenyl 3,4,6-tri-O-p-methoxybenzyl-1-thio-β-D-glucopyranoside (11). Following general process A, starting from 11a (50 mg, 0.1 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 11 as a white solid (32 mg, 52%): mp 102.3–104.6 °C; Rf = 0.33 (petroleum ether/ethyl acetate 6:1); 1H NMR (400 MHz, chloroform-d) δ 7.60–7.54 (m, 2H), 7.36–7.23 (m, 7H), 7.18–7.08 (m, 2H), 6.93–6.79 (m, 6H), 4.83 (s, 2H), 4.76 (d, J = 10.4 Hz, 1H), 4.57 (d, J = 11.6 Hz, 1H), 4.54–4.46 (m, 3H), 3.83 (s, 9H), 3.79–3.67 (m, 2H), 3.60–3.43 (m, 4H), 2.40 (d, J = 2.1 Hz, 1H) ppm. 13C NMR (100 MHz, chloroform-d) δ 159.34, 159.32, 159.19, 132.81, 131.96, 130.66, 130.35, 130.24, 129.65, 129.37, 128.95, 128.00, 113.97, 113.82, 113.76, 88.00, 85.64, 79.47, 77.12, 74.96, 74.69, 73.09, 68.62, 55.28, 43.68, 29.71, 14.63 ppm. [α]20D = −83.3 (c 0.09, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5S2Na+, 655.2342; found, 655.2326.
Phenyl 3,4,6-tri-O-benzyl-1-thio-β-D-galactopyranoside (12). Following general process A, starting from 12a (50 mg, 0.12 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 12 as a white solid (34.2 mg, 53%): mp 89.8–90.4 °C; Rf = 0.33 (petroleum ether/ethyl acetate 4:1); 1H NMR (400 MHz, chloroform-d) δ 7.59–7.48 (m, 2H), 7.41–7.07 (m, 18H), 4.89 (d, J = 11.5 Hz, 1H), 4.78–4.61 (m, 2H), 4.61–4.40 (m, 4H,H-1, ArCH2), 4.07–3.90 (m, 2H, H-2, H-4), 3.66 (s, 3H, H-5, H-6a and H-6b), 3.48 (dd, J = 9.3, 2.7 Hz, 1H, H-3), 2.46 (d, J = 2.2 Hz, 1H, OH) ppm. 13C NMR (100 MHz, chloroform-d) δ 138.64, 137.99, 137.85, 132.60, 132.21, 128.84, 128.56, 128.46, 128.19, 127.94, 127.88, 127.85, 127.75, 127.71, 127.58, 127.47, 88.51, 83.22, 77.62, 74.41, 73.61, 73.20, 72.43, 69.07, 68.70 ppm. [α]20D = −39.2 (c 0.13, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5S2Na+, 565.2025; found, 565.2014.
Phenyl 3,4-di-O-benzyl-6-O-tert-butyl-dimethylsily-1-thio-β-D-galactopyranoside (13). Following general process A, starting from 13a (50 mg, 0.114 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 13 as a colorless oil (38.6 mg, 60%). Rf = 0.45 (petroleum ether/ethyl acetate 8:1); 1H NMR (400 MHz, chloroform-d) δ 7.54–7.47 (m, 2H), 7.36–7.09 (m, 13H), 4.87 (d, J = 11.4 Hz, 1H), 4.68 (s, 2H), 4.57 (d, J = 11.4 Hz, 1H), 4.48 (d, J = 9.6 Hz, 1H), 4.01–3.93 (m, 1H), 3.91 (d, J = 2.7 Hz, 1H), 3.77–3.64 (m, 2H), 3.50–3.41 (m, 2H), 2.45–2.40 (m, 1H), 0.85 (s, 9H), 0.00 (s, 6H) ppm. 13C NMR (100 MHz, chloroform-d) δ 138.85, 138.11, 132.67, 132.14, 128.83, 128.56, 128.16, 127.89, 127.76, 127.59, 127.52, 127.38, 88.52, 83.31, 79.31, 74.43, 73.08, 72.53, 69.10, 61.52, 25.93, 18.23, −5.32, −5.42 ppm. [α]20D = +35.0 (c 0.1, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C32H42O5SiSNa+, 589.2420; found, 589.2396.
Phenyl 3-O-benzyl-4-O-acetyl-6-O-tert-butyl-dimethylsily-1-thio-β-D-galactopyranoside (14). Following general process A, starting from 14a (50 mg, 0.127 mmol), after 0.5 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 14 as a white solid (34.5 mg, 52%): mp 87.1–89.3 °C; Rf = 0.53 (petroleum ether/ethyl acetate 4:1); 1H NMR (400 MHz, chloroform-d) δ 7.55–7.48 (m, 2H), 7.32–7.21 (m, 8H), 5.56 (d, J = 3.0 Hz, 1H), 4.77 (d, J = 11.2 Hz, 1H), 4.55 (d, J = 9.7 Hz, 1H), 4.43 (d, J = 11.2 Hz, 1H), 3.73–3.65 (m, 2H), 3.64–3.54 (m, 2H), 3.46 (dd, J = 9.2, 3.1 Hz, 1H), 2.43 (d, J = 2.0 Hz, 1H), 2.04 (s, 3H), 0.84 (s, 9H), 0.00 (s, 6H) ppm. 13C NMR (100 MHz, chloroform-d) δ 170.05, 137.40, 132.64, 132.34, 128.87, 128.55, 128.27, 128.03, 127.82, 88.52, 80.45, 77.77, 71.73, 68.65, 65.98, 61.28, 25.81, 20.83, 18.21, −5.51, −5.61 ppm. [α]20D = −110 (c 0.03, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C27H38O6SiSNa+, 541.2056; found, 541.2047.
Phenyl 3,4-di-O-benzyl-1-thio-β-D-xyloside (15). Following general process A, starting from 15a (50 mg, 0.169 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 15 as colorless syrup (46.3 mg, 65%). Rf = 0.53 (petroleum ether/ethyl acetate 4:1); 1H NMR (400 MHz, chloroform-d) δ 7.54–7.48 (m, 2H), 7.41–7.19 (m, 13H), 4.92 (d, J = 5.9 Hz, 1H, H-1), 4.83 (d, J = 11.6 Hz, 1H), 4.74 (d, J = 11.6 Hz, 1H), 4.63 (s, 2H), 4.29 (dd, J = 11.7, 3.0 Hz, 1H, H-5b), 3.72 (q, J = 6.1 Hz, 1H, H-2), 3.63 (t, J = 6.1 Hz, 1H, H-3), 3.59–3.45 (m, 2H, H-4 and H-5a), 3.25 (d, J = 6.3 Hz, 1H, OH) ppm. 13C NMR (100 MHz, chloroform-d) δ 138.08, 137.56, 134.18, 131.91, 128.98, 128.57, 128.53, 128.05, 127.89, 127.83, 127.57, 88.95, 79.32, 77.27, 75.92, 73.89, 72.41, 70.83, 63.55 ppm. [α]20D = +85.0 (c 0.02, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C25H26O4SNa+, 445.1449; found, 445.1467.
Phenyl 2,
3,
3′,
4,
6,
6′-hexa-O-benzyl-D-1-thio-β-lactoside (16) [
32]. Following general process A, starting from
16a (100 mg, 0.118 mmol), after 1 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded
16 as colorless syrup (64.2 mg, 56%). Rf = 0.61 (petroleum ether/ethyl acetate 3:1);
1H NMR (400 MHz, chloroform-
d) δ 7.60 –7.55 (m, 2H), 7.41–7.15 (m, 33H), 5.07 (d,
J = 11.0 Hz, 1H), 4.96 (d,
J = 11.5 Hz, 1H), 4.85–4.73 (m, 2H), 4.73–4.62 (m, 3H), 4.58–4.47 (m, 3H), 4.48–4.37 (m, 2H), 4.34 (d,
J = 11.7 Hz, 1H), 4.26 (d,
J = 11.8 Hz, 1H), 3.98–3.89 (m, 2H), 3.88–3.73 (m, 3H), 3.62–3.54 (m, 1H), 3.53–3.35 (m, 6H), 2.50 (s, 1H) ppm.
13C NMR (100 MHz, chloroform-
d) δ 139.00, 138.80, 138.71, 138.47, 132.97, 132.00, 128.87, 128.41, 128.38, 128.27, 128.23, 128.17, 128.13, 127.91, 127.87, 127.75, 127.67, 127.57, 127.49, 127.45, 127.35, 102.86, 87.40, 84.18, 82.51, 79.98, 79.76, 76.06, 75.37, 75.03, 74.67, 73.60, 73.46, 73.10, 73.02, 72.65, 71.59, 68.34, 68.14 ppm. [α]
20D = −46.3 (c 0.08, CH
2Cl
2); HRMS (ESI-TOF) (
m/z): [M + Na]
+ calculated for C
60H
62O
10SNa
+, 997.3961; found, 997.3990.
4-Methylphenyl 3,
4,
6-tri-O-benzyl-1-thio-β-D-glucopyranoside (17) [
33]. Following general process A, starting from
2 (100 mg, 0.24 mmol), after 5 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded
17 as a white solid (99.2 mg, 74%). Rf = 0.51 (petroleum ether/ethyl acetate 6:1);
1H NMR (400 MHz, chloroform-
d) δ 7.50–7.41 (m, 2H), 7.40–7.15 (m, 15H), 7.10–6.97 (m, 2H), 4.91 (d,
J = 11.2 Hz, 1H), 4.87–4.78 (m, 2H), 4.65–4.50 (m, 3H), 4.43 (d,
J = 9.6 Hz, 1H), 3.81–3.68 (m, 2H), 3.61–3.55 (m, 2H), 3.54–3.49 (m, 1H), 3.49–3.40 (m, 1H), 2.40 (d,
J = 2.0 Hz, 1H), 2.31 (s, 3H) ppm.
4-Methoxyphenyl 3,
4,
6-tri-O-benzyl-1-thio-β-D-glucopyranoside (18) [
20]. Following general process A, starting from
2 (100 mg, 0.24 mmol), after 5 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded
18 as colorless syrup (105 mg, 76%). Rf = 0.63 (petroleum ether/ethyl acetate 8:1);
1H NMR (400 MHz, chloroform-
d) δ 7.60–7.46 (m, 2H), 7.41–7.29 (m, 13H), 7.25–7.13 (m, 2H), 6.82–6.70 (m, 2H), 4.96–4.80 (m, 3H), 4.68–4.53 (m, 3H), 4.39 (d,
J = 9.6 Hz, 1H), 3.80–3.77 (m, 5H), 3.62–3.50 (m, 3H), 3.42 (dd,
J = 9.6, 8.4 Hz, 1H), 2.43 (s, 1H) ppm.
4-Chlorophenyl 3,4,6-tri-O-benzyl-1-thio-β-D-glucopyranoside (19). Following general process A, starting from 2 (100 mg, 0.24 mmol), after 30 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 19 as colorless syrup (96 mg, 70%). Rf = 0.33 (petroleum ether/ethyl acetate 8:1); 1H NMR (400 MHz, chloroform-d) δ 7.56–7.49 (m, 2H), 7.42–7.18 (m, 17H), 4.94–4.79 (m, 3H), 4.64–4.54 (m, 3H), 4.49 (d, J = 9.6 Hz, 1H), 3.83–3.71 (m, 2H), 3.66–3.42 (m, 4H), 2.38 (d, J = 2.2 Hz, 1H) ppm. 13C NMR (100 MHz, chloroform-d) δ 138.38, 138.18, 137.97, 134.41, 134.32, 130.15, 129.10, 129.05, 128.81, 128.58, 128.47, 128.42, 128.00, 127.99, 127.91, 127.88, 127.70, 127.66, 87.61, 85.89, 79.35, 75.38, 75.09, 73.43, 72.44, 68.93, 38.00 ppm. [α]20D = −115 (c 0.04, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C33H33O5ClSNa+, 599.1635; found, 599.1611.
4-Aminophenyl 3,4,6-tri-O-benzyl-1-thio-β-D-glucopyranoside (20). Following general process A, starting from 2 (50 mg, 0.12 mmol), after 30 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 20 as colorless syrup (45 mg, 68%). Rf = 0.35 (petroleum ether/ethyl acetate 2:1); 1H NMR (600 MHz, CDCl3) δ 7.41–7.25 (m, 15H), 7.24–7.20 (m, 2H), 6.59–6.54 (m, 2H), 4.94 (d, J = 11.2 Hz, 1H), 4.88–4.81 (m, 2H), 4.66–4.54 (m, 3H), 4.33 (d, J = 9.6 Hz, 1H), 4.15 (q, J = 7.1 Hz, 1H), 3.82–3.74 (m, 2H), 3.64–3.54 (m, 2H), 3.42 (t, J = 9.0 Hz, 1H), 2.43 (s, 1H) ppm. 13C NMR (100 MHz, chloroform-d) δ 147.25, 138.57, 136.38, 128.48, 128.40, 128.33, 127.98, 127.76, 127.64, 127.49, 115.33, 88.26, 85.91, 79.46, 75.26, 75.06, 73.43, 72.21, 69.04, 29.71, 29.33 ppm. [α]20D = +61.5 (c 0.026, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5S2Na+, 580.2130; found, 580.2120.
Thiophen-2-ylthio 3,4,6-tri-O-benzyl-1-thio-β-D-glucopyranoside (21). Following general process A, starting from 2 (50 mg, 0.12 mol), after 30 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 21 as a red solid (41 mg, 63%): mp 90.9–92.1 °C; Rf = 0.52 (petroleum ether/ethyl acetate 5:1); 1H NMR (400 MHz, chloroform-d) δ 7.47–7.09 (m, 17H), 6.98 (dd, J = 5.5, 3.5 Hz, 1H), 4.92–4.76 (m, 3H), 4.70–4.51 (m, 3H), 4.30 (d, J = 9.4 Hz, 1H), 3.82–3.70 (m, 2H), 3.62–3.46 (m, 3H), 3.42 (ddd, J = 9.0, 6.2, 2.7 Hz, 1H), 2.36 (d, J = 2.4 Hz, 1H) ppm. 13C NMR (100 MHz, chloroform-d) δ 138.39, 138.04, 136.17, 131.08, 128.56, 128.43, 128.34, 128.00, 127.96, 127.88, 127.81, 127.63, 127.53, 87.53, 85.80, 79.69, 75.39, 75.06, 73.50, 71.86, 68.87, 29.39 ppm. [α]20D = −46.6 (c 0.058, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C31H32O5S2Na+, 571.1589; found, 571.1602.
Benzothiazol-2-yl 3,4,6-tri-O-benzyl-1-thio-β-D-glucopyranoside (22). Following general process A, starting from 2 (50 mg, 0.12 mmol), after 30 min of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded 22 as a white solid (51.3 mg, 72%): mp 119.9–123.0 °C; Rf = 0.36 (petroleum ether/ethyl acetate 6:1); 1H NMR (400 MHz, chloroform-d) δ 7.94 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.7 Hz, 1H), 7.39–7.23 (m, 14H), 7.23–7.17 (m, 2H), 5.07 (d, J = 9.5 Hz, 1H), 4.99–4.80 (m, 3H), 4.67–4.47 (m, 3H), 3.86–3.63 (m, 6H), 3.13 (d, J = 3.1 Hz, 1H) ppm. 13C NMR (100 MHz, chloroform-d) δ 211.54, 152.68, 138.40, 138.11, 138.00, 128.56, 128.45, 128.36, 128.01, 127.95, 127.89, 127.85, 127.76, 127.61, 126.31, 125.06, 122.43, 121.02, 86.49, 85.95, 79.94, 75.49, 75.10, 73.50, 68.65, 29.34, 14.14 ppm. [α]20D = −64.3 (c 0.07, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C34H33NO5S2Na+, 622.1698; found, 622.1707.
Phenyl 3,
4,
6-tri-O-benzyl-1-seleno-β-D-glucopyranoside (26) [
34]. Following general process A, starting from
2 (100 mg, 0.24 mmol), after 12 h of ring-opening reaction for the crude anhydro sugar, purification by silica gel flash column chromatography afforded
26 as a white solid (85.5 mg, 61%). Rf = 0.43 (petroleum ether/ethyl acetate 8:1);
1H NMR (400 MHz, chloroform-
d) δ 7.72–7.66 (m, 2H), 7.41–7.19 (m, 18H), 4.93 (d,
J = 11.2 Hz, 1H), 4.89–4.82 (m, 2H), 4.75 (d,
J = 9.7 Hz, 1H), 4.67–4.54 (m, 3H), 3.85–3.74 (m, 2H), 3.67–3.57 (m, 2H), 3.56–3.47 (m, 2H) ppm.
Phenyl 3,4,6-tri-O-benzyl-2-O-picolyl-β-D-1-thio-glucopyranoside (28). Following the general process B, starting from 2 (100 mg, 0.24 mmol), after 2 h of ring-opening reaction for the crude anhydro sugar and then reaction with PicBr•HBr (2.0 equiv) for 1 h in the presence of sodium hydride (6 equiv), the residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford 28 as a colorless syrup (101 mg, 66%). Rf = 0.33 (petroleum ether/ethyl acetate 4:1); 1H NMR (400 MHz, chloroform-d) δ 8.58–8.50 (m, 1H), 7.65 (td, J = 7.7, 1.9 Hz, 1H), 7.58–7.49 (m, 3H), 7.40–7.09 (m, 19H), 5.04 (d, J = 12.4 Hz, 1H), 4.96–4.75 (m, 4H), 4.69 (d, J = 9.6 Hz, 1H), 4.64–4.49 (m, 3H), 3.84–3.69 (m, 3H), 3.65 (t, J = 9.3 Hz, 1H), 3.55 (t, J = 9.1 Hz, 2H) ppm. 13C NMR (100 MHz, chloroform-d) δ 158.30, 149.00, 138.29, 138.20, 138.06, 136.52, 133.54, 132.04, 128.90, 128.44, 128.41, 128.35, 128.00, 127.92, 127.82, 127.70, 127.65, 127.56, 127.50, 122.35, 121.65, 87.19, 86.55, 81.32, 79.13, 75.93, 75.81, 75.06, 73.43, 69.04 ppm. [α]20D = −45 (c 0.04, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C39H39O5NSNa+, 656.2447; found, 656.2416.
Phenyl 3,4,6-tri-O-benzyl-2-O-picolyl-β-D-1-thio-galactopyranoside (29). Following the general process B, starting from 12a (100 mg, 0.24 mmol), after 2 h of ring-opening reaction for the crude anhydro sugar, and then reaction with PicBr•HBr (2.0 equiv) for 1 h in the presence of sodium hydride (6 equiv), the residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford 29 as a colorless syrup (73 mg, 48%). Rf = 0.35 (petroleum ether/ethyl acetate 4:1); 1H NMR (400 MHz, chloroform-d) δ 8.53 (d, J = 5.0 Hz, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.56–7.43 (m, 3H), 7.41–7.09 (m, 19H), 4.98–4.87 (m, 3H), 4.75–4.63 (m, 3H), 4.58 (d, J = 11.5 Hz, 1H), 4.51–4.37 (m, 2H), 4.01–3.90 (m, 2H), 3.72–3.57 (m, 4H) ppm. 13C NMR (100 MHz, chloroform-d) δ 158.67, 148.91, 138.75, 138.14, 137.89, 136.40, 133.94, 131.56, 128.80, 128.46, 128.39, 128.22, 127.96, 127.86, 127.84, 127.64, 127.49, 127.11, 122.24, 121.79, 87.52, 83.91, 78.04, 77.40, 77.34, 77.08, 76.76, 76.28, 74.49, 73.62, 73.48, 72.57, 68.79 ppm. [α]20D = +9.1 (c 0.33, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C39H39O5NSNa+, 656.2447; found, 656.2433.
Phenyl 3,
4,
6-Tri-O-benzyl-2-O-(phenylmethoxy)methyl-β-D-1-thio-glucopyranoside (30) [
21]. Following the general process B, starting from
2 (100 mg, 0.24 mmol), after 2 h of ring-opening reaction for the crude anhydro sugar and then reaction with BOMCl (2.0 equiv) for 3 h in the presence of sodium hydride (6 equiv), the residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford
30 as a white solid (100.3 mg, 63%). Rf = 0.53 (petroleum ether/ethyl acetate 6:1);
1H NMR (400 MHz, CDCl
3) δ 7.59–7.49 (m, 2H), 7.39–7.12 (m, 23H), 5.06 (d,
J = 6.4 Hz, 1H), 4.94 (d,
J = 6.4 Hz, 1H), 4.92 –4.83 (m, 3H), 4.79 (d,
J = 10.8 Hz, 1H), 4.69–4.58 (m, 3H), 4.58–4.49 (m, 2H), 3.77 (dd,
J = 10.9, 2.0 Hz, 1H), 3.74–3.61 (m, 4H), 3.52 (m, 1H) ppm.
Phenyl 3,
4,
6-Tri-O-benzyl-2-O-(cyanomethyl)-β-D-1-thio-glucopyranoside (31) [
18]. Following the general process B, starting from
2 (100 mg, 0.24 mmol), after 2 h of ring-opening reaction for the crude anhydro sugar and then reaction with bromoacetonitrile (2.5 equiv) for 2 h in the presence of sodium hydride (6 equiv), the residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford
31 as a white solid (76.8 mg, 55%). Rf = 0.53 (petroleum ether/ethyl acetate 6:1);
1H NMR (400 MHz, CDCl
3) δ 7.61–7.52 (m, 2H), 7.41–7.22 (m, 16H), 7.19 (dd,
J = 7.2, 2.3 Hz, 2H), 4.90–4.82 (m, 2H), 4.80 (d,
J = 10.9 Hz, 1H), 4.64–4.56 (m, 2H), 4.55 (s, 1H), 4.52 (t,
J = 1.8 Hz, 1H), 4.51–4.39 (m, 2H), 3.77 (dd,
J = 10.9, 2.1 Hz, 1H), 3.72 (dd,
J = 10.9, 4.3 Hz, 1H), 3.67–3.62 (m, 2H), 3.47 (m, 1H), 3.35 (m, 1H) ppm.
Phenyl 3,
4,
6-Tri-O-benzyl-2-O-(2-cyanobenzyl)-β-D-1-thio-glucopyranoside (32) [
22]. Following the general process B, starting from
2 (100 mg, 0.24 mmol), after 2 h of ring-opening reaction for the crude anhydro sugar and then reaction with 2-cyanobenzyl bromide (2.0 equiv) for 1 h in the presence of sodium hydride (6 equiv), the residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford
32 as a colorless oil (107.4 mg, 68%). Rf = 0.31 (petroleum ether/ethyl acetate 6:1);
1H NMR (400 MHz, CDCl
3) δ 7.73–7.53 (m, 6H), 7.43–7.19 (m, 18H), 5.11 (d,
J = 12.5 Hz, 1H), 5.03 (d,
J = 12.5 Hz, 1H), 4.91–4.79 (m, 3H), 4.73–4.56 (m, 4H), 3.83 (dd,
J = 10.9, 2.0 Hz, 1H), 3.78 (d,
J = 4.4 Hz, 1H), 3.77–3.66 (m, 2H), 3.60–3.51 (m, 2H) ppm.
Phenyl 3,
4,
6-Tri-O-benzyl-2-O-benzoyl-β-D-1-thio-glucopyranoside (33) [
35]. Following the general process B, starting from
2 (100 mg, 0.24 mmol), after 2 h of ring-opening reaction for the crude anhydro sugar and then reaction with BzCl (3.0 equiv) for 2 h in the presence of sodium hydride (6 equiv), the residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford
33 as a white solid (97.8 mg, 63%). Rf = 0.61 (petroleum ether/ethyl acetate 6:1);
1H NMR (400 MHz, CDCl
3) δ 8.11–8.03 (m, 2H), 7.60 (d,
J = 7.4 Hz, 1H), 7.54–7.44 (m, 4H), 7.43–7.20 (m, 13H), 7.18–7.11 (m, 5H), 5.31 (dd,
J = 10.0, 9.0 Hz, 1H), 4.87–4.78 (m, 2H), 4.75 (d,
J = 11.0 Hz, 1H), 4.70–4.55 (m, 4H), 3.91–3.82 (m, 2H), 3.81–3.74 (m, 2H), 3.64 (m, 1H) ppm.
Phenyl 3,
4,
6-Tri-O-benzyl-2-O-pivaloyl-β-D-1-thio-glucopyranoside (34) [
36]. Following the general process B, starting from
2 (100 mg, 0.24 mmol), after 2 h of ring-opening reaction for the crude anhydro sugar and then reaction with PivCl (3.0 equiv) for 2 h in the presence of sodium hydride (6 equiv), the residue was purified by column chromatography on silica gel (ethyl acetate-hexane gradient elution) to afford
34 as a white solid (78.3 mg, 52%). Rf = 0.73 (petroleum ether/ethyl acetate 8:1);
1H NMR (400 MHz, CDCl
3) δ 7.58–7.49 (m, 2H), 7.41–7.22 (m, 16H), 7.19 (dd,
J = 7.2, 2.4 Hz, 2H), 5.13 (t, 1H), 4.84–4.76 (m, 2H), 4.75–4.53 (m, 5H), 3.81 (dd, 1 H, J = 1.5, 11.0 Hz, 1H), 3.79–3.67 (m, 3H), 3.59 (m, 1H), 1.26 (s, 9H) ppm.
Methyl 2,3,4-tri-O-benzoyl-6-O-(3,4,6-tri-O-benzyl-2-O-picolyl-β-D-glucopyranosyl)-α-D-glucopyranoside (35). Following the general process C, the glycosylation between 28 (100 mg, 0.16 mmol, 1.3 equiv) and methyl 2,3,4-tri-O-benzoyl-α-D-glucopyranoside (1.0 equiv) led to 30. Purification by silica gel flash column chromatography afforded 35 as a colorless syrup (87.5 mg, 70%, β-only). Rf = 0.26 (petroleum ether/ethyl acetate 2:1); 1H NMR (400 MHz, chloroform-d) δ 8.53 (d, J = 4.9 Hz, 1H), 8.03–7.76 (m, 7H), 7.66–7.58 (m, 1H), 7.55–7.46 (m, 3H), 7.45–7.20 (m, 19H), 7.17–7.07 (m, 3H), 6.14 (t, J = 9.8 Hz, 1H), 5.42 (t, J = 9.9 Hz, 1H), 5.24–5.16 (m, 2H), 5.11 (d, J = 3.6 Hz, 1H), 4.95–4.83 (m, 2H), 4.82–4.73 (m, 2H), 4.56–4.46 (m, 3H), 4.44 (d, J = 12.3 Hz, 1H), 4.34 (td, J = 8.7, 8.2, 4.1 Hz, 1H), 4.10 (dd, J = 10.9, 2.1 Hz, 1H), 3.77 (dd, J = 11.1, 7.8 Hz, 1H), 3.73–3.57 (m, 4H), 3.53–3.41 (m, 2H), 3.34 (s, 3H) ppm. 13C NMR (100 MHz, chloroform-d) δ 165.81, 165.74, 165.50, 158.78, 148.95, 138.50, 138.14, 138.09, 136.46, 133.42, 133.33, 133.05, 129.93, 129.90, 129.66, 129.28, 129.11, 128.90, 128.41, 128.34, 128.25, 127.98, 127.94, 127.75, 127.58, 122.18, 121.45, 103.94, 96.68, 84.47, 82.78, 77.65, 75.68, 75.40, 75.00, 74.95, 73.45, 72.11, 70.49, 69.91, 68.97, 68.61, 55.52 ppm. [α] 20D = +40.9 (c 0.22, CH2Cl2); HRMS (ESI-TOF) (m/z): [M + Na]+ calculated for C61H59O14NNa+, 1052.3833 ; found, 1052.3814.
Methyl 2,
3,
4-tri-O-benzyl-6-O-(3,
4,
6-tri-O-benzyl-2-O-picolyl-β-D-glucopyranosyl)-α-D-glucopyranoside (36) [
37]. Following the general process C, the glycosylation between
28 (100 mg, 0.16 mmol, 1.3 equiv) and methyl 2,3,4-tri-
O-benzyl-α-D-glucopyranoside (1.0 equiv) led to
36. Purification by silica gel flash column chromatography afforded
36 as a colorless syrup (97.7 mg, 82%, β-only). Rf = 0.46 (petroleum ether/ethyl acetate 2:1);
1H NMR (400 MHz, chloroform-
d) δ 8.44 (d,
J = 4.8 Hz, 1H), 7.48–7.20 (m, 28H), 7.19–7.11 (m, 4H), 7.02 (t,
J = 6.1 Hz, 1H), 5.13 (d,
J = 12.9 Hz, 1H), 4.96–4.85 (m, 3H), 4.84–4.70 (m, 4H), 4.68–4.59 (m, 2H), 4.58–4.51 (m, 4H), 4.47 (d,
J = 11.0 Hz, 1H), 4.39 (d,
J = 7.8 Hz, 1H), 4.17 (d,
J = 10.7 Hz, 1H), 3.95 (t,
J = 9.3 Hz, 1H), 3.78 (dt,
J = 14.3, 7.1 Hz, 1H), 3.72–3.61 (m, 4H), 3.60–3.40 (m, 5H), 3.31 (s, 3H) ppm.
Methyl 2,
3,
6-tri-O-benzyl-4-O-(3,
4,
6-tri-O-benzyl-2-O-picolyl-β-D-glucopyranosyl)-α-D-glucopyranoside (37) [
37]. Following the general process C, the glycosylation between
28 (100 mg, 0.16 mmol, 1.3 equiv) and methyl 2,3,6-tri-
O-benzyl-α-D-glucopyranoside (1.0 equiv) led to
37. Purification by silica gel flash column chromatography afforded
37 as a colorless syrup (59.6 mg, 50%, β-only). Rf = 0.48 (petroleum ether/ethyl acetate 2:1);
1H NMR (400 MHz, chloroform-
d) δ 8.50 (d,
J = 4.8 Hz, 1H), 7.54 (t,
J = 7.7 Hz, 1H), 7.45–7.06 (m, 32H), 5.07 (d,
J = 11.3 Hz, 1H), 4.96 (d,
J = 13.5 Hz, 1H), 4.90–4.71 (m, 6H), 4.64–4.52 (m, 4H), 4.48–4.34 (m, 4H), 3.96 (t,
J = 9.5 Hz, 1H), 3.87–3.77 (m, 2H), 3.72 (d,
J = 10.9 Hz, 1H), 3.65–3.57 (m, 2H), 3.48 (m, 5H), 3.34 (s, 3H), 3.31–3.27 (m, 1H) ppm.
Methyl 2,
4,
6-tri-O-benzyl-3-O-(3,
4,
6-tri-O-benzyl-2-O-picolyl-β-D-glucopyranosyl)-α-D-glucopyranoside (38) [
37]. Following the general process C, the glycosylation between
28 (100 mg, 0.16 mmol, 1.3 equiv) and methyl 2,4,6-tri-
O-benzyl-α-D-glucopyranoside (1.0 equiv) led to
38. Purification by silica gel flash column chromatography afforded
38 as a colorless syrup (95.4 mg, 80%, β-only). Rf = 0.65 (petroleum ether/ethyl acetate 2:1);
1H NMR (400 MHz, chloroform-
d) δ 8.56 (d,
J = 4.9 Hz, 1H), 7.56–7.47 (m, 2H), 7.40–7.04 (m, 31H), 5.29 (d,
J = 13.5 Hz, 1H), 5.14–4.95 (m, 4H), 4.89 (d,
J = 10.9 Hz, 1H), 4.83 (d,
J = 10.6 Hz, 1H), 4.70–4.53 (m, 3H), 4.53–4.33 (m, 7H), 3.81–3.64 (m, 6H), 3.63–3.48 (m, 4H), 3.45 (d,
J = 9.4 Hz, 1H), 3.30 (s, 3H) ppm.
Methyl 3,
4,
6-tri-O-benzyl-2-O-(3,
4,
6-tri-O-benzyl-2-O-picolyl-β-D-glucopyranosyl)-α-D-glucopyranoside (39) [
37]. Following the general process C, the glycosylation between
28 (100 mg, 0.16 mmol, 1.3 equiv) and methyl 3,4,6-tri-
O-benzyl-α-D-glucopyranoside led to
39. Purification by silica gel flash column chromatography afforded
39 as a colorless syrup (91.8 mg, 77%, β-only). Rf = 0.55 (petroleum ether/ethyl acetate 2:1);
1H NMR (400 MHz, chloroform-
d) δ 8.42 (d,
J = 5.0 Hz, 1H), 7.45–7.10 (m, 30H), 7.11–6.96 (m, 3H), 5.18 (d,
J = 13.4 Hz, 1H), 5.03–4.85 (m, 3H), 4.85–4.70 (m, 4H), 4.67–4.56 (m, 3H), 4.55–4.40 (m, 4H), 4.01 (t,
J = 9.2 Hz, 1H), 3.89–3.61 (m, 10H), 3.58 (t,
J = 7.4 Hz, 1H), 3.43 (m, 1H), 3.39 (s, 3H) ppm.
1,
2:5,
6-Di-O-isopropylidine-3-O-(3,
4,
6-tri-O-benzyl-2-O-picolyl-β-D-glucopyranosyl)-α-D-glucofuranose (40) [
37]. Following the general process C, the glycosylation between
28 (100 mg, 0.16 mmol, 1.3 equiv) and 1,2:5,6-bis-
O-(1-methylethylidene)-α-D-glucofuranose (1.0 equiv) led to
40. Purification by silica gel flash column chromatography afforded
40 as colorless syrup (49.2 mg, 52%, β-only). Rf = 0.35 (petroleum ether/ethyl acetate 2:1);
1H NMR (400 MHz, chloroform-
d) δ 8.56 (d,
J = 4.9 Hz, 1H), 7.67–7.56 (m, 1H), 7.43–7.22 (m, 14H), 7.21–7.08 (m, 3H), 5.81 (d,
J = 3.7 Hz, 1H), 4.93–4.86 (m, 2H), 4.85–4.75 (m, 3H), 4.66–4.47 (m, 5H), 4.43 (q,
J = 5.9 Hz, 1H), 4.38–4.28 (m, 2H), 4.12–4.01 (m, 2H), 3.78–3.58 (m, 4H), 3.47–3.38 (m, 2H), 1.48 (s, 3H), 1.41 (s, 3H), 1.31 (s, 3H), 1.25 (s, 3H) ppm.
Methyl 2,
3,
4-tri-O-benzyl-6-O-(3,
4,
6-tri-O-benzyl-2-O-picolyl-β-D-galactopyranosyl)-α-D-glucopyranoside (41) [
37]. Following the general process C, the glycosylation between
29 (100 mg, 0.16 mmol, 1.3 equiv) and methyl 2,3,4-tri-
O-benzyl-α-D-glucopyranoside (1.0 equiv) led to
41. Purification by silica gel flash column chromatography afforded
41 as a colorless syrup (102.5 mg, 86%, β-only). Rf = 0.36 (petroleum ether/ethyl acetate 2:1);
1H NMR (400 MHz, chloroform-
d) δ 8.43 (d,
J = 4.8 Hz, 1H), 7.48–7.39 (m, 2H), 7.39–7.08 (m, 30H), 7.04 (q,
J = 4.7 Hz, 1H), 5.09 (d,
J = 13.2 Hz, 1H), 4.98–4.87 (m, 3H), 4.80–4.38 (m, 11H), 4.35 (d,
J = 7.7 Hz, 1H), 4.13 (d,
J = 10.8 Hz, 1H), 3.97–3.84 (m, 3H), 3.79 (dd,
J = 10.5, 5.0 Hz, 1H), 3.52 (m, 7H), 3.28 (s, 3H) ppm.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
